Introduction
Several clinical trials demonstrate that addition of LABA rather than increasing the dose of ICS shows beneficial effects with regards to symptom control and pulmonary function. Beclomethasone dipropionate (BDP), a widely used ICS has a favourable risk/efficacy profile and has been recently developed as an extra-fine formulation with hydrofluoroalkane (HFA) propellant, showing proven efficacy in controlling asthma symptoms in both adults and children at a 2.5-fold lower daily dose compared to chlorofluorocarbon-BDP. The extra-fine formulation of an ICS is mainly designed for a uniform drug delivery to both central and peripheral airways, consistent treatment of airway inflammation throughout the lower respiratory tract. A reduced ICS dose implies a lower systemic exposure and a lower overall risk of steroid-specific side-effects. A recently developed technology uses the HFA-134a propellant to obtain an extra-fine formulation of new drugs, as well as reformulation of pre-existing drugs in a pressurised metered-dose inhaler (pMDI).
|
Addition of a long-acting b2-agonist (LABA) to a low-to-medium dose of inhaled corticosteroids (ICS) is highly recommended by international guidelines in asthma patients inadequately controlled on ICS monotherapy. |
Aim
The present study aimed to assess the efficacy and tolerability and establish the non-inferiority of equipotent doses of the combination of Beclomethasone dipropionate-formoterol (BDP/F) pMDI when compared with those of a budesonide-formoterol (BUD/F) dry powder inhaler (DPI) in patients with moderate to severe asthma whose symptoms were not controlled with ICS alone.
Method
Patient Profile
Inclusion Criteria
The study included adult patients of 18–65 yrs with moderate-to-severe persistent asthma and with a forced expiratory volume in one second (FEV1) 50–80% of predicted normal values. All patients underwent initial treatment with ICS at a daily dose of 1,000 mg of BDP-equivalent and had inadequately controlled asthma symptoms occurring more than once a week.
Exclusion Criteria
Patients with chronic obstructive pulmonary disease; current or ex-smokers; severe asthma exacerbation or symptomatic infection of the airways in the previous 8 weeks; three or more courses of oral corticosteroids or hospitalisation due to asthma in the previous 6 months; treatment with LABAs, anticholinergics or antihistamines in the previous 2 weeks, and/or with topical or intra-nasal corticosteroids and leukotriene antagonists in the previous 4 weeks; and change of ICS dose in the previous 4 weeks were excluded from the study.
Study Design
The present study was a phase III, multinational, multicentre, double-blind, double-dummy, randomised, two-arm parallel groups, controlled study design. 219 patients after a 2-week run-in period with moderate-to-severe asthma were randomised, to a 12-week treatment with BDP/F 200 mg+12 mg b.i.d. delivered via a pMDI or BUD/F 400 mg+12 mg b.i.d. delivered via a DPI.
Endpoints
Primary Endpoints
Morning pre-dose peak expiratory flow (PEF) measured by patients, at least 12 h after the previous evening dose, in the last 2 weeks of the treatment period (weeks 11–12). Pulmonary function tests (PFT) namely FEV1, forced vital capacity (FVC), PEF and mid-expiratory flow at 50% vital capacity (MEF50%) were performed at each visit before study. Occurrence of asthma exacerbations categorised as mild, moderate and severe were evaluated at all post-baseline visits.
Secondary Endpoints
Tolerability profile was the secondary outcome.
Statistical analysis
The sample size calculation was made by defining the limit for non-inferiority as the lower limit of the unilateral 97.5% confidence interval (CI) for the difference between least square means (LSMs) of morning PEF being ≥-20 L.min-1.
Results
The two groups matched well for baseline characteristics in terms of demographics, pulmonary function, symptoms and time from first asthma diagnosis. A comparable baseline asthma severity profile was observed between the two groups and no significant differences were observed between groups in mean ICS dose.
Efficacy profile analysis
Lung function
Primary outcome: Morning pre-dose PEF during the last 2 weeks of the treatment period, the difference between adjusted means (LSMs) of the BDP/F group (338.3 L.min-1) and the BUD/F group (337.8 L.min-1) was 0.49 L.min-1. Both treatment groups showed marked and significant improvements in morning PEF; the mean increases from baseline to end-point were 29.43¡52.8 L.min-1 (95% CI: 19.31–39.54) and 28.63¡43.4 L.min-1 (20.39– 36.87) in the BDP/F and BUD/F groups, respectively (fig. 1).
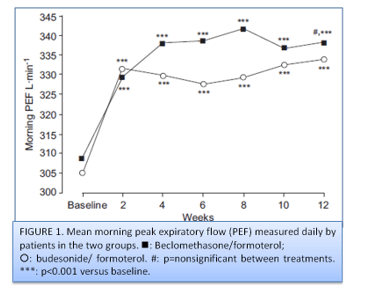
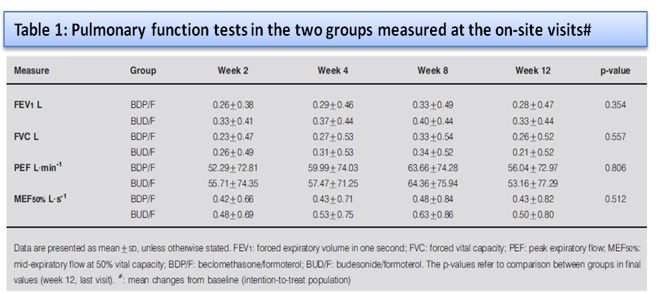
No significant difference was observed between the two groups for evening PEF at the end of treatment. Compared to baseline, a significant increase was observed in daily FEV1 measured by patients in both groups. The results of the PFT measured at the on-site visits (FEV1, FVC, PEF and MEF50%) are shown in table 1 and figure 2.
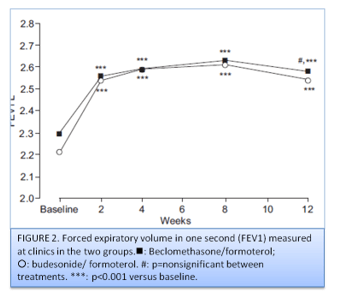
Statistically significant improvements from baseline were found in both groups from week 2 onwards in all lung function parameters.
Symptoms
Both groups showed a significant decrease from baseline for clinical symptoms’ scores from the first 2-week period, as well as daily use of rescue salbutamol, with no significant difference between groups at treatment completion. From baseline to end-point (weeks 11–12) in daytime the mean decrease in the symptoms’ scores were -0.93±0.78 U and -0.86±0.86 U in the BDP/F and BUD/F groups, respectively (p<0.001 versus baseline). During night-time the mean changes observed from baseline to the end of treatment for the symptoms’ score were -0.73±0.75 U and -0.66±0.84 U in the BDP/F and BUD/F groups, respectively (p<0.001 versus baseline). A significant decrease was observed for daily use of rescue medication from 2.16±1.15 puffs.day-1 in the last week of run-in to 0.76±0.92 puffs/day-1 in the last 2 weeks of treatment period in the BDP/F group, and from 2.28±1.50 to 0.87±1.04 puffs.day-1 in the BUD/F group.
Exacerbations
29 patients; 17 (15.9%) in the BDP/F group and 12 (11.0%) in the BUD/F group manifested asthma exacerbations. No severe exacerbations were noted. In both groups two patients reported with moderate exacerbations requiring one course of oral corticosteroids. No statistically significant difference was observed in the ratio of days of exacerbation to days of exposure was 0.013¡0.04 in the BDP/F group and 0.023¡0.11 in the BUD/F group (p50.38). For the BDP/F group the first exacerbation was observed after 29 days and was 24 (1–69) days for the BUD/F group (p50.342 between groups in the Kaplan–Meier estimate for survival curves).
Tolerability
The two treatment groups reported no significant differences. 15 (13.8%) patients in the BDP/F group and 18 (16.5%) in the BUD/F group reported adverse effect profile due to likely due to seasonal affections and not drug tolerability. None of the groups reported changes in ECG or QTc interval prolongation or change in blood pressure during the study period.
|
Previous studies have reported a better drug delivery profile of fine particle dose of BUD, i.e. the amount of drug supposed to reach the lower airways after one inhalation of DPI combination 200/6 mg BUD/F, is 46.0 mg, compared to the fine particle dose of BDP, after one inhalation of extra-fine 100/6 mg BDP/F, reported as 34.5 mg. |
Conclusions
A significant decrease in the use of rescue medications and an equivalent beneficial effect in lung function and in clinical symptoms was demonstrated by the study results for the two tested combinations. Rates of asthma exacerbation and/or the need of additional prevention therapy did not report any significant differences between the groups. Thus, BDP/F combination was as efficacious as BUD/F for treatment of asthma.
Adapted from: Papi A, Paggiaro PL, Nicolini G, Vignola AM, Fabbri LM. Beclomethasone/formoterol versus budesonide/formoterol combination therapy in asthma. European Respiratory Journal. 2007 Apr 1;29(4):682-9.