Each vial contains:
Bivalirudin .. 250 mg
Excipients .q.s.
Supplied with 5 ml Sterile Water for Injection I.P



Each vial contains:
Bivalirudin .. 250 mg
Excipients .q.s.
Supplied with 5 ml Sterile Water for Injection I.P
Sterile lyophilized powder to be reconstituted, for single use
Mechanism of action
Bivalirudin directly inhibits thrombin by specifically binding both to the catalytic site and to the anion-binding exosite of circulating and clot-bound thrombin. Thrombin is a serine proteinase that plays a central role in the thrombotic process, acting to cleave fibrinogen into fibrin monomers and to activate Factor XIII to Factor XIIIa, allowing fibrin to develop a covalently cross-linked framework which stabilizes the thrombus; thrombin also activates Factors V and VIII, promoting further thrombin generation, and activates platelets, stimulating aggregation and granule release. The binding of bivalirudin to thrombin is reversible as thrombin slowly cleaves the bivalirudin-Arg3-Pro4 bond, resulting in recovery of thrombin active site functions.
In in vitro studies, bivalirudin inhibited both soluble (free) and clot-bound thrombin, was not neutralized by products of the platelet release reaction, and prolonged the activated partial thromboplastin time (aPTT), thrombin time (TT), and prothrombin time (PT) of normal human plasma in a concentration-dependent manner. The clinical relevance of these findings is unknown.
Anticoagulant effect
In healthy volunteers and patients (with ≥70% vessel occlusion undergoing routine percutaneous transluminal coronary angiography [PTCA]), bivalirudin exhibited dose- and concentration-dependent anticoagulant activity as evidenced by prolongation of the activated clotting time (ACT), aPTT, PT, and TT. Intravenous administration of, bivalirudin produces an immediate anticoagulant effect. Coagulation times return to baseline approximately 1 hour following cessation of bivalirudin administration.
In 291 patients with ≥70% vessel occlusion undergoing routine PTCA, a positive correlation was observed between the dose of bivalirudin and the proportion of patients achieving ACT values of 300 sec or 350 sec. At a bivalirudin dose of 1 mg/kg IV bolus plus 2.5 mg/kg/h IV infusion for 4 hours, followed by 0.2 mg/kg/h, all patients reached maximal ACT values >300 sec.
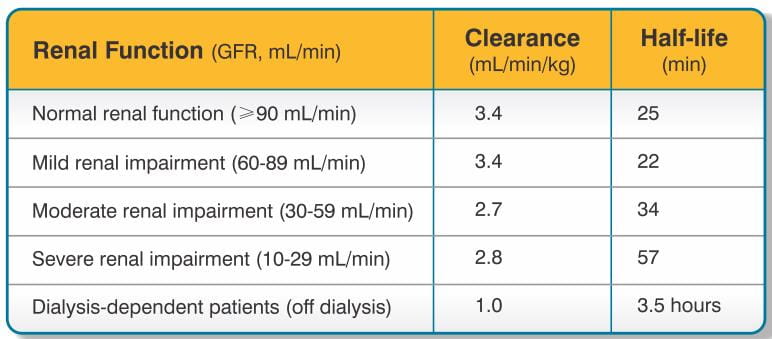
Bivalirudin exhibits linear pharmacokinetics following IV administration to patients undergoing PTCA. In these patients, a mean steady state bivalirudin concentration of 12.3 ± 1.7 mcg/mL is achieved following an IV bolus of 1 mg/kg and a 4-hour 2.5 mg/kg/h IV infusion. Bivalirudin does not bind to plasma proteins (other than thrombin) or to red blood cells. Bivalirudin is cleared from plasma by a combination of renal mechanisms and proteolytic cleavage, with a half-life in patients with normal renal function of 25 min.
The disposition of bivalirudin was studied in PTCA patients with mild, moderate, and severe renal impairment. Drug elimination was related to glomerular filtration rate (GFR). Total body clearance was similar for patients with normal renal function and with mild renal impairment (60-89 mL/min). Clearance was reduced in patients with moderate and severe renal impairment and in dialysis-dependent patients (See Table 1 for pharmacokinetic parameters).
Bivalirudin is hemodialyzable, with approximately 25% cleared by hemodialysis.
This was a randomized, double-blind, multicenter study involving 6002 (intent-to-treat) patients undergoing percutaneous coronary intervention (PCI). Patients were randomized to treatment with bivalirudin with the "provisional" use of platelet glycoprotein IIb/IIIa inhibitor (GPI) or heparin plus planned use of GPI. GPIs were added on a "provisional" basis to patients who were randomized to bivalirudin in the following circumstances:
During the study, one or more of these circumstances occurred in 12.7% of patients in the bivalirudin with provisional GPI arm. GPIs were administered to 7.2% of patients in the bivalirudin with provisional GPI arm (62.2% of eligible patients).
Patients ranged in age from 25-95 years (median, 63); weight ranged from 35-199 kg (median 85.5); 74.4% were male and 25.6% were female. Indications for PCI included unstable angina (35% of patients), myocardial infarction within 7 days prior to intervention (8% of patients), stable angina (25%) and positive ischemic stress test (24%). Stents were deployed in 85% of patients. Ninety-nine percent of patients received aspirin and 86% received thienopyridines prior to study treatment.
Bivalirudin was administered as a 0.75 mg/kg bolus followed by a 1.75 mg/kg/h infusion for the duration of the procedure. The activated clotting time (ACT) was measured 5 min after the first bolus of study medication. If the ACT was <225 seconds, an additional bolus of 0.3 mg/kg was given. At investigator discretion, the infusion could be continued following the procedure for up to 4 hours. The median infusion duration was 44 min. Heparin was administered as a 65 U/kg bolus. The ACT was measured 5 min after the first bolus of study medication. If the ACT was <225 seconds, an additional bolus of 20 units/kg was given. GPIs (either abciximab or eptifibatide) were given according to manufacturers' instructions. Both randomized groups could be given "provisional" treatments during the PCI at investigator discretion, but under double-blind conditions. "Provisional" treatment with GPI was requested in 5.2% of patients randomized to heparin plus GPI (they were given placebo) and 7.2% patients randomized to bivalirudin with provisional GPI (they were given abciximab or eptifibatide according to pre-randomization investigator choice and patient stratification).
The percent of patients reaching protocol-specified levels of anticoagulation was greater in the bivalirudin with provisional GPI group than in the heparin plus GPI group. For patients randomized to bivalirudin with provisional GPI, the median 5 min ACT was 358 sec (interquartile range 320-400 sec) and the ACT was <225 sec in 3%. For patients randomized to heparin plus GPI, the median 5 min ACT was 317 sec (interquartile range 263-373 sec) and the ACT was <225 sec in 12%. At the end of the procedure, median ACT values were 334 sec (bivalirudin group) and 276 sec (heparin plus GPI group).
For the composite endpoint of death, MI, or urgent revascularization adjudicated under double-blind conditions, the frequency was higher (7.6%) (95% confidence interval 6.7%-8.6%) in the bivalirudin with "provisional" GPI arm when compared to the heparin plus GPI arm (7.1%) (95% confidence interval 6.1%-8.0%). However, major hemorrhage was reported significantly less frequently in the bivalirudin with provisional GPI arm (2.4%) compared to the heparin plus GPI arm (4.1%). Study outcomes are shown in Table 2.
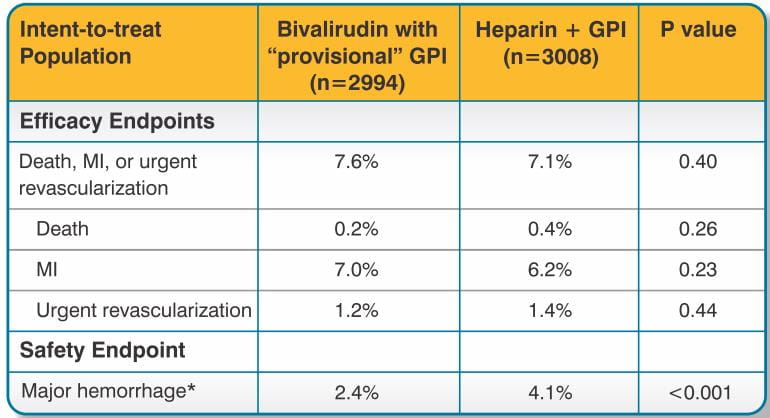
At 12 months follow-up, mortality was 1.9% among patients randomized to bivalirudin with "provisional" GPIs and 2.5% among patients randomized to heparin plus GPI.
Bivalirudin was evaluated in patients with unstable angina undergoing PTCA in two randomized, double-blind, multicenter studies with identical protocols. Patients must have had unstable angina defined as: (1) a new onset of severe or accelerated angina or rest pain within the month prior to study entry or (2) angina or ischemic rest pain which developed between four hours and two weeks after an acute myocardial infarction (MI). Overall, 4312 patients with unstable angina, including 741 (17%) patients with post-MI angina, were treated in a 1:1 randomized fashion with bivalirudin or heparin. Patients ranged in age from 29-90 (median 63) years, their weight was a median of 80 kg (39-120 kg) and 68% were male. Twenty-three percent of patients were treated with heparin within one hour prior to randomization. All patients were administered aspirin 300-325 mg prior to PTCA and daily thereafter. Patients randomized to bivalirudin were started on an intravenous infusion of bivalirudin (2.5 mg/kg/h). Within 5 min after starting the infusion, and prior to PTCA, a 1 mg/kg loading dose was administered as an intravenous bolus. The infusion was continued for 4 hours, then the infusion was changed under double-blinded conditions to bivalirudin (0.2 mg/kg/h) for up to an additional 20 hours (patients received this infusion for an average of 14 hours). The ACT was checked at 5 min and at 45 min following commencement. If on either occasion the ACT was <350 sec, an additional double-blinded bolus of placebo was administered. The bivalirudin dose was not titrated to ACT. Median ACT values were: ACT in sec (5th percentile-95th percentile): 345 sec (240-595 sec) at 5 min and 346 sec (range 269-583 sec) at 45 min after initiation of dosing. Patients randomized to heparin were given a loading dose (175 IU/kg) as an intravenous bolus 5 min before the planned procedure, with immediate commencement of an infusion of heparin (15 IU/kg/h). The infusion was continued for 4 hours. After 4 hours of infusion, the heparin infusion was changed under double-blinded conditions to heparin (15 IU/kg/h) for up to 20 additional hours. The ACT was checked at 5 min and at 45 min following commencement. If on either occasion the ACT was <350 sec, an additional double-blind bolus of heparin (60 IU/kg) was administered. Once the target ACT was achieved for heparin patients, no further ACT measurements were performed. The protocol allowed use of open-label heparin at the discretion of the investigator after discontinuation of blinded study medication, whether or not an endpoint event (procedural failure) had occurred. The use of open-label heparin was similar between bivalirudin and heparin treatment groups (about 20% in both groups).
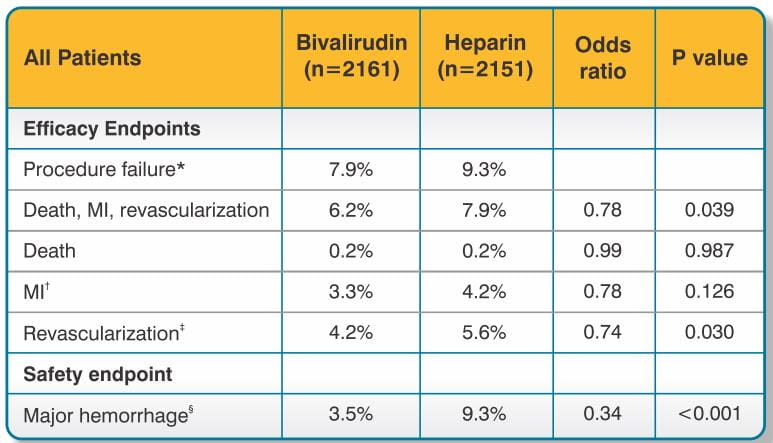
The studies were designed to demonstrate the safety and efficacy of bivalirudin in patients undergoing PTCA as a treatment for unstable angina as compared with a control group of similar patients receiving heparin during and up to 24 hours after initiation of PTCA. The primary protocol endpoint was a composite endpoint called procedural failure, which included both clinical and angiographic elements measured during hospitalization. The clinical elements were: the occurrence of death, MI, or urgent revascularization, adjudicated under double-blind conditions. The angiographic elements were: impending or abrupt vessel closure. The protocol-specified safety endpoint was major hemorrhage.
The median duration of hospitalization was 4 days for both the bivalirudin and the heparin treatment groups. The rates of procedural failure were similar in the bivalirudin and heparin treatment groups. Study outcomes are shown in Table 3.
The ACUITY trial was a prospective, randomised open-label, trial of bivalirudin with or without GP IIb/IIIa inhibitor (Arms B and C respectively) versus unfractionated heparin or enoxaparin with GP IIb/IIIa inhibitor (Arm A) in 13,819 high risk ACS patients.
In Arms B and C of the ACUITY trial, the recommended dose of bivalirudin was an initial post-randomisation IV bolus of 0.1 mg/kg followed by a continuous IV infusion of 0.25 mg/kg/h during angiography or as clinically warranted.
For patients undergoing PCI, an additional IV bolus of 0.5 mg/kg bivalirudin was administered and the rate of IV infusion increased to 1.75 mg/kg/h.
In Arm A of the ACUITY trial, UFH or enoxaparin was administered in accordance with the relevant guidelines for the management of ACS in patients with UA and NSTEMI. Patients in Arms A and B were also randomised to receive a GP IIb/IIIa inhibitor either upfront at the time of randomization (prior to angiography) or at the time of PCI. A total of 356 (7.7%) of patients randomised to Arm C also received a GP IIb/IIIa inhibitor.
High risk patient characteristics of the ACUITY population that mandated angiography within 72 hours were balanced across the three treatment arms. Approximately 77% of patients had recurrent ischaemia, approximately 70% had dynamic ECG changes or elevated cardiac biomarkers, approximately 28% had diabetes and approximately 99% of patients underwent angiography within 72 hours.
Following angiographic assessment, patients were triaged to either medical management (33%), PCI (56%) or CABG (11%). Additional anti-platelet therapy utilized in the study included aspirin and clopidogrel.
The primary analysis and results for ACUITY at 30-days and 1 year for the overall (ITT) population is shown in Tables 4. The advantage of bivalirudin over UFH/enoxaparin plus GP IIb/IIIa inhibitor in terms of bleeding events was only observed in the bivalirudin monotherapy arm.
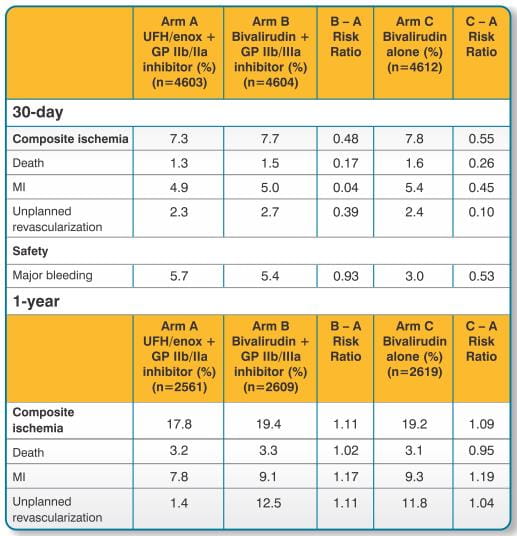
The HORIZONS trial was a prospective, dual arm, single blind, randomised, multi-centre trial to establish the safety and efficacy of bivalirudin in patients with STEMI undergoing a primary PCI strategy with stent implantation with either a slow release paclitaxal-eluding stent or an otherwise identical uncoated bare metal stent. A total of 3,602 patients were randomized to receive either bivalirudin (1,800 patients) or unfractionated heparin plus a GP IIb/IIIa inhibitor (1,802 patients). All patients received aspirin and clopidogrel with twice as many patients (approximately 64%) receiving a 600mg loading dose of clopidogrel than a 300mg loading dose of clopidogrel. Approximately 66% of patients were pre-treated with unfractionated heparin.
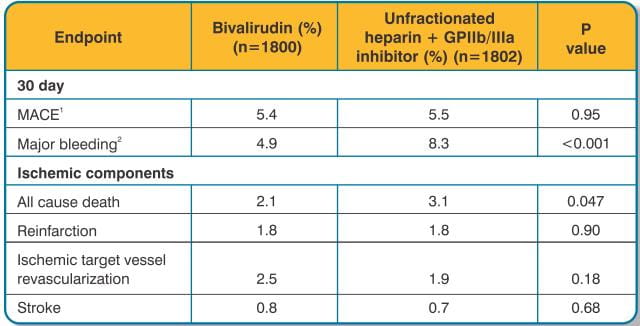
The dose of bivalirudin used in HORIZONS was the same as that used in the REPLACE-2 study (0.75 mg/kg bolus followed by a 1.75 mg/kg body weight/hour infusion). A total of 92.9% of patients treated underwent primary PCI as their primary management strategy.
The analysis and results for the HORIZONS trial at 30 days for the overall population is shown in Table 5. Results at 1 year were consistent with results at 30 days.
This was a single-group open-label study which enrolled 51 patients with heparin-induced thrombocytopenia (HIT) or heparin induced thrombocytopenia and thrombosis syndrome (HITTS) undergoing PCI. Evidence for the diagnosis of HIT/HITTS was based on a clinical history of a decrease of platelets in patients after heparin administration [new diagnosis or history of clinically suspected or objectively documented HIT/HITTS defined as either: 1) HIT: positive heparin-induced platelet aggregation (HIPA) or other functional assay where the platelet count has decreased to <100,000/mL (minimum 30% from prior to heparin), or has decreased to <150,000/mL (minimum 40% from prior to heparin), or has decreased as above within hours of receiving heparin in a patient with a recent, previous exposure to heparin; 2) HITTS: thrombocytopenia as above plus arterial or venous thrombosis diagnosed by physician examination/laboratory and/or appropriate imaging studies]. Patients ranged in age from 48-89 years (median 70); weight ranged from 42-123 kg (median 76); 50% were male and 50% were female. Bivalirudin was administered as either 1 mg/kg bolus followed by 2.5 mg/kg/h (high dose in 28 patients) or 0.75 mg/kg bolus followed by a 1.75 mg/kg/h infusion (lower dose in 25 patients) for up to 4 hours. Ninety-eight percent of patients received aspirin, 86% received clopidogrel and 19% received GPIs.
The median ACT values at the time of device activation were 379 sec (high dose) and 317 sec (lower dose). Following the procedure, 48 of the 51 patients (94%) had TIMI grade 3 flow and stenosis <50%. One patient died during a bradycardic episode 46 hours after successful PCI, another patient required surgical revascularization, and one patient experienced no flow requiring a temporary intra-aortic balloon.
Two of the fifty-one patients with the diagnosis of HIT/HITTS developed thrombocytopenia after receiving bivalirudin and GPIs.
BIVASTAT is indicated for use as an anticoagulant in patients with unstable angina undergoing PTCA.
BIVASTAT with provisional use of GPI is indicated for use as an anticoagulant in patients undergoing PCI.
BIVASTAT is indicated for patients with, or at risk of HIT or HITTS undergoing PCI.
BIVASTAT in these indications is intended for use with aspirin and has been studied only in patients receiving concomitant aspirin.
The safety and effectiveness of bivalirudin have not been established in patients with acute coronary syndromes who are not undergoing PTCA or PCI.
BIVASTAT is for intravenous administration only.
Bivalirudin is intended for use with aspirin (300-325 mg daily) and has been studied only in patients receiving concomitant aspirin.
For Patients Who do not have HIT/HITTS
The recommended dose of BIVASTAT is an intravenous (IV) bolus dose of 0.75 mg/kg, followed by an infusion of 1.75 mg/kg/h for the duration of the PCI/PTCA procedure. Five min after the bolus dose has been administered, an ACT should be performed and an additional bolus of 0.3 mg/kg should be given if needed.
Provisional use of GPI administration should be considered in following circumstances:
For Patients Who have HIT/HITTS
The recommended dose of BIVASTAT in patients with HIT/HITTS undergoing PCI is an IV bolus of 0.75 mg/kg. This should be followed by a continuous infusion at a rate of 1.75 mg/kg/h for the duration of the procedure.
For Ongoing Treatment Post Procedure
Continuation of the BIVASTAT infusion following PCI/PTCA for up to 4 hours post-procedure is optional, at the discretion of the treating physician. After four hours, an additional IV infusion of BIVASTAT may be initiated at a rate of 0.2 mg/kg/h (low-rate infusion), for up to 20 hours, if needed.
No reduction in the bolus dose is needed for any degree of renal impairment. The infusion dose of BIVASTAT may need to be reduced, and anticoagulant status monitored in patients with renal impairment. Patients with moderate renal impairment (30-59 mL/min) should receive an infusion of 1.75 mg/kg/h. If the creatinine clearance is less than 30 mL/min, reduction of the infusion rate to 1 mg/kg/h should be considered. If a patient is on hemodialysis, the infusion rate should be reduced to 0.25 mg/kg/h.
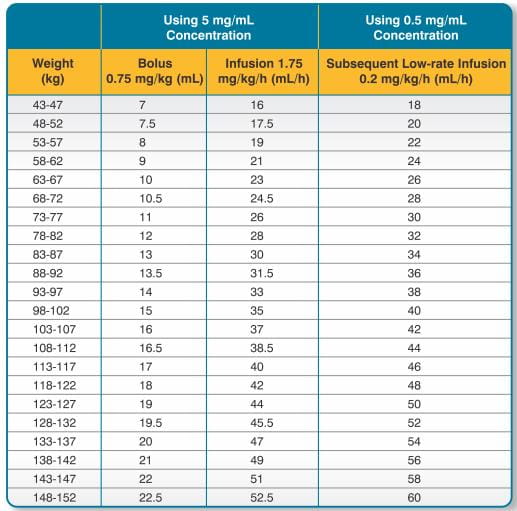
BIVASTAT is intended for intravenous bolus injection and continuous infusion after reconstitution and dilution. To each 250 mg vial, add 5 mL of Sterile Water for Injection, I.P. Gently swirl until all material is dissolved. Each reconstituted vial should be further diluted in 50 mL of 5% Dextrose in Water or 0.9% Sodium Chloride for Injection to yield a final concentration of 5 mg/mL (e.g., 1 vial in 50 mL; 2 vials in 100 mL; 5 vials in 250 mL). The dose to be administered is adjusted according to the patient's weight (See Table 6).
If the low-rate infusion is used after the initial infusion, a lower concentration bag should be prepared. In order to prepare this bag, reconstitute the 250 mg vial with 5 mL of Sterile Water for Injection, I.P. Gently swirl until all material is dissolved. Each reconstituted vial should be further diluted in 500 mL of 5% Dextrose in Water or 0.9% Sodium Chloride for Injection to yield a final concentration of 0.5 mg/mL. The infusion rate to be administered should be selected from the right-hand column in Table 6.
BIVASTAT should be administered via an intravenous line. No incompatibilities for bivalirudin have been observed with glass bottles or polyvinyl chloride bags and administration sets. The following drugs should not be administered in the same intravenous line with BIVASTAT, since they resulted in haze formation, microparticulate formation, or gross precipitation when mixed with bivalirudin: alteplase, amiodarone HCl, amphotericin B, chlorpromazine HCl, diazepam, prochlorperazine edisylate, reteplase, streptokinase, and vancomycin HCl. Dobutamine was compatible at concentrations up to 4 mg/mL but incompatible at a concentration of 12.5 mg/mL.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration. Preparations of BIVASTAT containing particulate matter should not be used. Reconstituted material will be a clear to slightly opalescent, colorless to slightly yellow solution.
Do not freeze reconstituted or diluted BIVASTAT. Reconstituted material may be stored at 2-8 C for up to 24 hours. Diluted BIVASTAT with a concentration of between 0.5 mg/mL and 5 mg/mL is stable at room temperature for up to 24 hours. Discard any unused portion of reconstituted solution remaining in the vial
.In clinical trials in patients undergoing PCI/PTCA, co-administration of bivalirudin with heparin, warfarin, thrombolytics, or GPIs was associated with increased risks of major bleeding events compared to patients not receiving these concomitant medications.
There is no experience with co-administration of bivalirudin and plasma expanders such as dextran.
Although most bleeding associated with the use of bivalirudin in PCI/PTCA occurs at the site of arterial puncture, hemorrhage can occur at any site. An unexplained fall in blood pressure or hematocrit should lead to serious consideration of a hemorrhagic event and cessation of bivalirudin administration. Bivalirudin should be used with caution in patients with disease states associated with an increased risk of bleeding.
An increased risk of thrombus formation, including fatal outcomes, has been associated with the use of bivalirudin in gamma brachytherapy.
If a decision is made to use bivalirudin during brachytherapy procedures, maintain meticulous catheter technique, with frequent aspiration and flushing, paying special attention to minimizing conditions of stasis within the catheter or vessels.
The disposition of bivalirudin was studied in PTCA patients with mild, moderate and severe renal impairment. The clearance of bivalirudin was reduced approximately 20% in patients with moderate and severe renal impairment and was reduced approximately 80% in dialysis-dependent patients. The infusion dose of bivalirudin may need to be reduced, and anticoagulant status monitored in patients with renal impairment (see Dosage and Administration).
Category B
Reproductive studies have been performed in rats at subcutaneous doses up to 150 mg/kg/day, (1.6 times the maximum recommended human dose based on body surface area) and rabbits at subcutaneous doses up to 150 mg/kg/day (3.2 times the maximum recommended human dose based on body surface area). These studies revealed no evidence of impaired fertility or harm to the fetus attributable to bivalirudin. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Bivalirudin is intended for use with aspirin. Because of possible adverse effects on the neonate and the potential for increased maternal bleeding, particularly during the third trimester, bivalirudin and aspirin should be used together during pregnancy only if clearly needed.
It is not known whether bivalirudin is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when BIVASTAT is administered to a nursing woman.
The safety and effectiveness of bivalirudin in pediatric patients have not been established
In studies of patients undergoing PCI, 44% were ≥65 years of age and 12% of patients were ≥75 years old. Elderly patients experienced more bleeding events than younger patients. Patients treated with bivalirudin experienced fewer bleeding events in each age stratum, compared to heparin.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
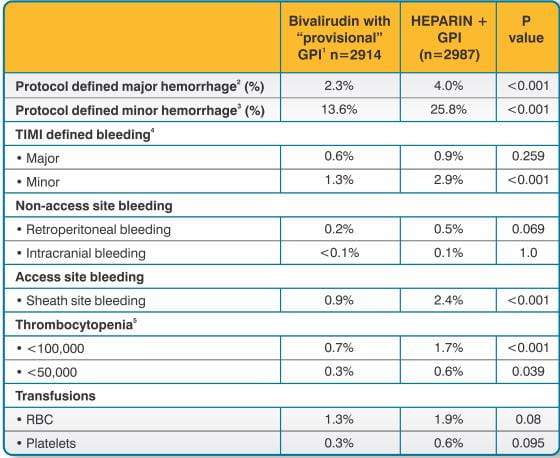
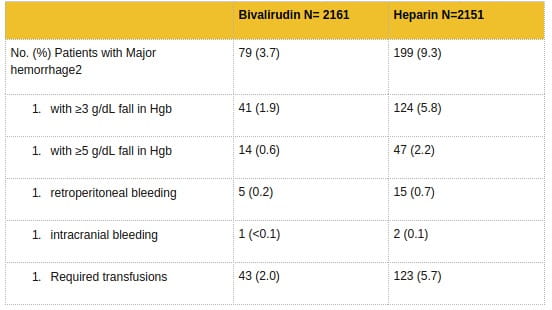
In 6010 patients undergoing PCI treated in the REPLACE-2 trial, bivalirudin patients exhibited statistically significantly lower rates of bleeding, transfusions, and thrombocytopenia as noted in Table 7.
In 4312 patients undergoing PTCA for treatment of unstable angina in 2 randomized, double-blind studies comparing bivalirudin to heparin, bivalirudin patients exhibited lower rates of major bleeding and lower requirements for blood transfusions. The incidence of major bleeding is presented in Table 8. The incidence of major bleeding was lower in the bivalirudin group than in the heparin group.
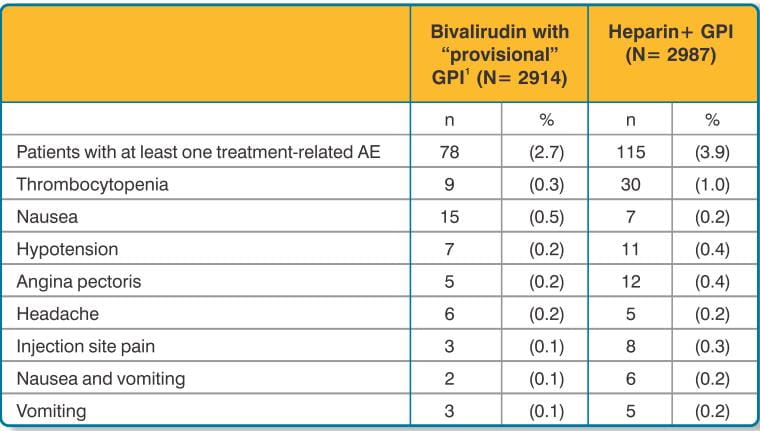
In the AT-BAT study, of the 51 patients with HIT/HITTS, 1 patient who did not undergo PCI had major bleeding during CABG on the day following angiography. Nine patients had minor bleeding (mostly due to access site bleeding), and 2 patients developed thrombocytopenia.
Adverse reactions, other than bleeding, observed in clinical trials were similar between the bivalirudin treated patients and the control groups.
Adverse reactions (related adverse events) seen in clinical studies in patients undergoing PCI and PTCA are shown in Tables 9 and 10.
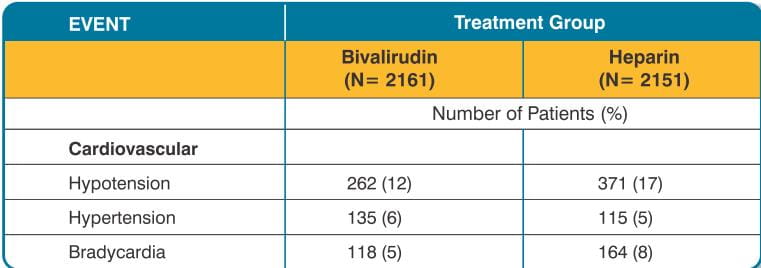
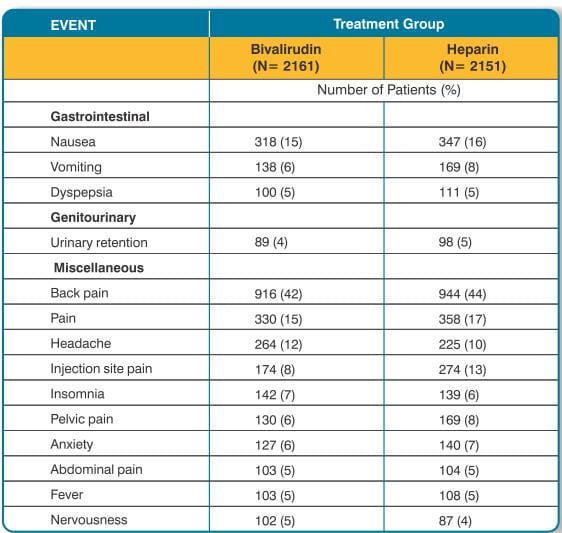
Serious, non-bleeding adverse events were experienced in 2% of 2161 bivalirudin- treated patients and 2% of 2151 heparin-treated patients. The following individual serious non-bleeding adverse events were rare (>0.1% to <1%) and similar in incidence between bivalirudin- and heparin-treated patients. These events are listed by body system:
Body as a Whole: fever, infection, sepsis;
Cardiovascular: hypotension, syncope, vascular anomaly, ventricular fibrillation; Nervous: cerebral ischemia, confusion, facial paralysis;
Respiratory: lung edema;
Urogenital: kidney failure, oliguria.
In the BAT trial, there was no causality assessment for adverse events.
In in vitro studies, bivalirudin exhibited no platelet aggregation response against sera from patients with a history of HIT/HITTS.
Among 494 subjects who received bivalirudin in clinical trials and were tested for antibodies, 2 subjects had treatment-emergent positive bivalirudin antibody tests. Neither subject demonstrated clinical evidence of allergic or anaphylactic reactions and repeat testing was not performed. Nine additional patients who had initial positive tests were negative on repeat testing.
Because postmarketing adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been identified during postapproval use of bivalirudin: fatal bleeding; hypersensitivity and allergic reactions including reports of anaphylaxis; lack of anticoagulant effect; thrombus formation during PCI with and without intracoronary brachytherapy, including reports of fatal outcomes.
Cases of overdose of up to 10 times the recommended bolus or continuous infusion dose of bivalirudin have been reported in clinical trials and in post-marketing reports. A number of the reported overdoses were due to failure to adjust the infusion dose of bivalirudin in persons with renal dysfunction including persons on hemodialysis. Bleeding, as well as deaths due to hemorrhage, have been observed in some reports of overdose. In cases of suspected overdosage, discontinue bivalirudin immediately and monitor the patient closely for signs of bleeding. There is no antidote to bivalirudin. Bivalirudin is hemodialyzable.
BIVASTAT I.V. Injection 250mg ..Vial of 10 ml supplied with 5 ml of Sterile Water for Injection I.P.