Desirox
Product Monograph
Patients with chronic anaemias such as thalassaemia, sickle cell disease, congenital rare anaemias and myelodysplastic syndromes require regular blood transfusions in order to improve both quality of life and survival. Humans are unable to eliminate the iron released from the breakdown of transfused red blood cells and the excess iron is deposited as haemosiderin and ferritin in the liver, spleen, endocrine organs and myocardium. The accumulation of toxic quantities of iron causes tissue damage and leads to complications such as heart failure, diabetes, hypothyroidism and liver failure. Morbidity and mortality in regularly transfused thalassaemia patients are due primarily to the effects of iron overload rather than to the underlying disease.
Iron chelators mobilize tissue iron by forming soluble, stable complexes that are then excreted in the faeces and/or urine. Although desferrioxamine has been available for the past 40 years, its poor oral bioavailability and short plasma half-life necessitate parenteral administration and prolonged infusions. Further, therapy was expensive and cumbersome, and led to significant non-compliance.
The introduction of the world's first oral iron chelator, Kelfer (deferiprone), by Cipla in 1995 revolutionised the treatment of iron overload, and brought the benefits of oral iron chelation therapy within reach of thousands of patients globally.
Deferasirox is yet another breakthrough in oral iron chelation therapy. Its once-daily dosing and availability as a dispersible tablet are unique features, and represent an important advance in therapy. This monograph reviews the available literature on this promising molecule.
Iron accumulation can occur either through excessive absorption in the gastrointestinal tract (primary haemochromatosis) or exogenous administration via multiple transfusions of red blood cells. Normal body iron stores in the typical patient are 3 - 4 g; an excess of iron of 20 g or more can lead to organ damage. Each unit of transfused red blood cells averages 200 - 250 mg of iron.11
Patients with hereditary and refractory anaemias require frequent blood transfusions as part of ongoing therapy (Table 1). The most common of these conditions worldwide are sickle cell disease and ß-thalassaemia. Each year, about 210,000 infants are born with sickle cell disease and about 90,000 are born with ß-thalassaemia. In many countries of the Mediterranean, Middle East and Asia, ß-thalassaemia major is the most common haemoglobin disorder.1 In India, about 10,000 babies are born with thalassaemia per year.

Regular transfusions are the mainstay of ß-thalassaemia major treatment. The addition of regular blood transfusions to the treatment of patients with sickle cell disease is becoming increasingly common for specific complications, such as stroke prevention and the treatment of acute chest syndrome.1
The process of iron metabolism in the human body has been well described. In a state of metabolic balance, the human body absorbs 1 to 2 mg of iron daily from diet and loses a similar amount through cellular sloughing. 12 Physiological mechanisms of iron excretion from the body via bile, urine, faeces and perspiration are extremely limited. 3
Iron overload, or haemosiderosis, is a complication of the chronic blood transfusions used to treat several haematological disorders, including ß-thalassaemia major, sickle cell disease and myelodysplastic syndrome. Iron is efficiently conserved within the body, and signs of iron overload may be noted after transfusion of 10-20 units of packed red blood cells, delivering approximately 2-5 g of elemental iron. Paradoxically, gastrointestinal absorption of iron is also abnormally increased in these conditions, further contributing to total iron stores.13 Most of the iron released to the circulation is derived from the catabolism of thalassaemic RBCs.14
Excess iron is depicted in the form of haemosiderins (insoluble iron deposits) in the visceral tissues, thereby progressively damaging the cardiac, hepatic and endocrine systems. In addition, once the capacity of transferrin to bind iron is surpassed, further organ damage can result from a highly toxic unbound iron moiety (non-transferrin-bound iron or NTBI). NTBI can catalyze the formation of reactive hydroxyl radicals. 13 Subsequently, peroxidative damage to proteins and membrane lipids occurs.2
The consequences of untreated iron overload include hepatotoxicity, hypogonadism, diabetes mellitus, osteopenia and cardiac hypertrophy. Cardiac dysfunction accounts for most of the early mortality in patients with iron overload.13
In patients with ß-thalassaemia major, transfusional chronic iron overload complications most frequently associated with mortality are those related to myocardial dysfunction. 1 Cardiac damage can lead to arrhythmias, congestive heart failure and death.5 Other common complications seen in these patients include hepatic fibrosis, liver cirrhosis, diabetes mellitus, osteoporosis and impaired growth and development in children. 1 There is some correlation between organ iron concentrations and organ damage. 2
The toxic side effects of transfusional iron overload can be ameliorated using chelating drugs, which can remove iron, maintain negative iron balance (iron excretion > iron absorption plus iron from transfusions) and low, safe iron levels in organs, most importantly the heart.
6
In iron overload, the hepatocytes are the major cells of iron storage, with liver iron closely approximating total body iron.
11 Threshold values of 15 mg/g dry liver weight and 2500 mg/L serum ferritin have been established as indicators of increased risk of cardiac complications. Direct assessment of myocardial iron may offer a valuable additional parameter.
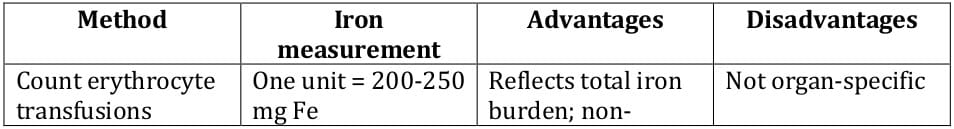
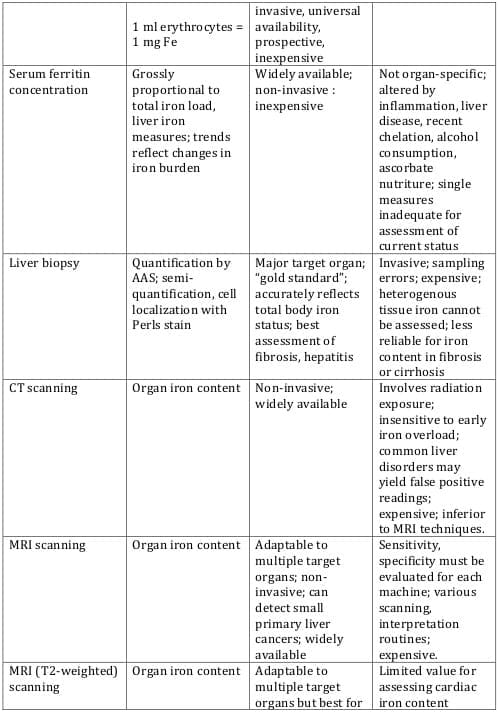
9 Table 2 lists traditional as well as newer approaches for iron estimation, with their advantages and disadvantages.
Chelation therapy is generally recommended after 20 units of erythrocytes or if serum ferritin increases to > 1000 µg/L.
1
The benefit from iron chelation by reducing morbidity and mortality in patients with iron overload from regular red blood cell transfusions, especially in thalassaemia, has been demonstrated over the past 40 years in innumerable studies. 4
The introduction of desferrioxamine in the 1960s for the treatment of transfusional siderosis has changed the life expectancy and life quality of patients with thalassaemia major. Studies have shown a cohort-of-birth related improvement in survival, reflected in an inverse, mirror-like decrease in cardiac mortality, supporting the claim that prevention of cardiac mortality is the most important beneficial effect of chelation therapy. In former years, the course of established myocardial disease in transfusional haemosiderosis was uniformly fatal. Unfortunately, compliance with the vigorous requirements of daily subcutaneous infusions is still a serious limiting factor in treatment outcome. 9
More recent studies with the oral iron chelator deferiprone have shown the following:
- Long-term deferiprone treatment provides greater protection than does desferrioxamine against the cardiotoxicity of iron.16
- Conventional treatment with desferrioxamine did not prevent excess cardiac iron accumulation in more than half the patients with thalassaemia major. Oral deferiprone was more effective at removing cardiac iron.17
- After one year of treatment, the improvement in T2* and increase in left ventricular ejection fraction were significantly greater in patients treated with deferiprone than those receiving desferrioxamine. The authors concluded that deferiprone monotherapy was significantly more effective than desferrioxamine in improving asymptomatic myocardial siderosis in ?-thalassaemia major.18
- In another study that evaluated the effect of deferiprone on cardiac mortality, 157 patients received deferiprone for a median duration of 4.3 years between 1995 and the end of 2003. 359 patients received desferrioxamine.
There were 52 cardiac events including ten cardiac deaths among patients treated with desferrioxamine. In contrast, no cardiac event occurred in any of the patients during deferiprone treatment or within 18 months after deferiprone. The estimated hazard of a cardiac event on deferiprone was less than one-tenth that in patients on desferrioxamine.19
In clinical practice, a key goal for chelation therapy is the prevention of iron accumulation, as this will prevent a rise in liver iron concentration and the secondary redistribution of iron to other organs including the pituitary and endocrine glands and the heart.8 The excretion of iron in either urine or faeces must keep pace with the rate of iron accumulated from blood transfusion. This requirement in thalassaemia major is on average about 0.5 mg/kg/day but is actually remarkably variable (from 0.32 to 0.64 mg/kg/day) and with even higher requirements in some hypersplenic patients.8 Hepatic iron content of 2.7 mg/g dry weight liver is a goal of chelation therapy.11
A second important concept in chelation therapy is “protection time” against forms of potentially damaging iron species, either in the plasma as non-transferrin-bound iron, or in cells as labile iron. Although the majority of cellular iron is stored as soluble ferritin or insoluble haemosiderin, this iron is available neither for rapid chelation nor for participation in potentially damaging redox cycling of iron. The tiny fraction of cellular iron that is available for iron chelation is often referred to as the chelatable iron pool or the labile iron pool. Because these iron pools are rapidly (re)incorporated into ferritin and only transiently available for chelation, the efficiency of chelation therapy and its iron- detoxifying effect depend upon the drug being available 24 hours a day. 8
The ideal iron chelator
The ideal properties of an effective chelating agent are: 2, 8, 9
- Selective affinity for ferric iron (Fe3+), the stable oxidized form of iron found in plasma and stores.
- Drug availability 24 hours a day, as efficiency of chelation depends on this
- Oral activity
- Ability to remove iron from the liver, myocardium and other tissues
- Lack of toxicity in the kidneys and bone marrow
- Limited central nervous system or foetal access.
Mechanism of action
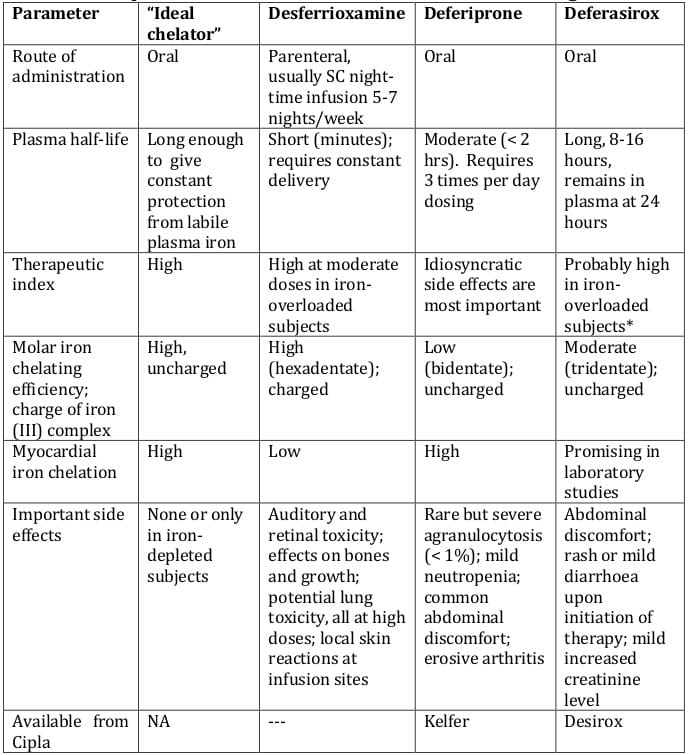
Ferric iron has six coordination sites, which need to be chelated completely to prevent the generation of free radicals. Chelators such as desferrioxamine coordinate all six sites using a single molecule (hexadentate chelator). Chelators such as deferiprone possess two coordination sites (bidentate),7 requiring 3 molecules to form a complete complex with ferric iron (Table 3).
Bidentate and tridentate ligands are generally well absorbed via the gastrointestinal tract, unlike hexadenate ligands, because of their smaller size.3

Currently available iron chelators include desferrioxamine, deferiprone and more recently, deferasirox (Table 4).
Desferrioxamine
Desferrioxamine (deferoxamine) has been in use since 1963. 7 It is an effective treatment and has prolonged the lifespan of adequately chelated patients.1 However, its delivery is less than ideal. It is administered as a subcutaneous infusion over 8 to 24 hours 5 to 7 days per week, and is most commonly delivered overnight by a small portable pump.2 In a significant proportion of patients, the burdensome administration requirements and poor patient compliance have limited its therapeutic efficacy.1 It is estimated that up to one-third of patients who are prescribed desferrioxamine do not adhere to therapy.2 It is also expensive.
As a result, there is a need for an economical, effective and easily administered iron chelator, as most patients require life-long chelation therapy.1
Deferiprone
Deferiprone (Kelfer) is the world's first iron oral chelator, introduced by Cipla in India in1995. Subsequently, it was approved by the European Union in 1999.
It has demonstrated efficacy in reducing iron overload, particularly of the myocardium. 1 Iron-related cardiac disease is a major clinical problem. The liver can be readily unloaded by aggressive chelation much more rapidly than can the heart. 10
Further, deferasirox and deferiprone are more effective in entering cells and chelating intracellular iron than desferrioxamine.9 Lastly, deferiprone is relatively affordable.
Deferasirox
In November 2005, the US FDA approved deferasirox under an accelerated programme permitting the use of treatments for serious and life-threatening diseases based on early demonstrations of efficacy.1
With a plasma half-life of 8 to 16 hours, once-daily dosing permits circulating drug at all times to scavenge non-transferrin-bound “labile plasma iron,” which is the chemical species responsible for tissue damage in iron-overloaded subjects.10
It is an orally administered once-daily treatment. The pharmacology, efficacy and safety of this new iron chelator are described in detail in the following chapters.
Deferasirox is an N-substituted bis-hydroxyphenyl-triazole3
Empiric formula of iron-free ligand: C21H15N3O4.
Deferasirox is highly selective for Fe3+ over Fe2+ and has minimal affinity for other divalent metal ions, such as zinc or copper. 1
Being a tridentate ligand, two molecules of deferasirox are required to coordinate one Fe3+, with each deferasirox molecule providing three of the six co-ordinating sites required for complete coordination of Fe3+ (Figure 1). The 2:1 ligand-to-iron complex is the form found under physiological conditions. The three coordinating sites on each molecule are provided by one nitrogen atom and two oxygen atoms. 8
Biochemical properties
Deferasirox mobilises tissue iron by forming soluble, stable complexes that are then excreted in the faeces. Iron is chelated both from the reticuloendothelial cells as well as various parenchymal tissues. The chelated iron is cleared by the liver and excreted through the bile. It also has the ability to prevent myocardial cell iron uptake and remove iron directly from myocardial cells. In animal models, on a molar basis, it has been shown to be 5 times more potent than desferrioxamine and 10 times more potent than deferiprone. Iron removal also depends on plasma concentration, host factors, degree of loading, and rate of accessibility of stored iron to chelator.20
Efficacy of chelation
The theoretical efficacy of chelation of deferasirox (the proportion of excreted iron relative to the amount that could theoretically have been bound) was 15.3, 20.5 and 15.6% in the 10-, 20- and 40-mg/kg groups, respectively. 3
Pharmacodynamic properties
The pharmacodynamic properties of deferasirox in patients with transfusional iron overload have been evaluated using three key parameters:
- iron balance (iron excretion : iron intake)
- myocardial iron levels
- labile plasma iron (LPI)
I. Iron Balance Studies
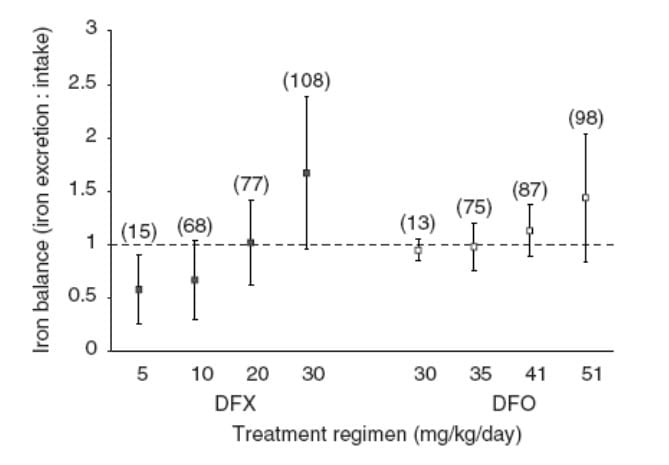
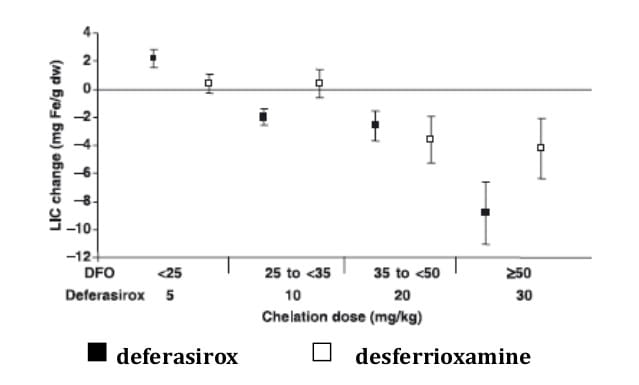
These studies examine the ratio of iron excretion to iron intake. In the phase III clinical trial comparing deferasirox with desferrioxamine, net iron balance (iron excretion : iron intake ≈ 1) or net iron excretion (iron excretion : iron intake > 1) was achieved in 69% of patients (Figure 2). These patients had higher baseline liver iron concentrations (LIC) of > 7 mg Fe/g dry weight (dw) and were treated with high dosages of either study drug (deferasirox ≥ 20 mg/kg/day, desferrioxamine > 41 mg/kg/day). The 31% of patients who did not achieve net iron balance or net iron excretion had baseline LIC ≤ 7 mg Fe/g dw and were treated with deferasirox < 20 mg/kg/day or desferrioxamine < 41 mg/kg/day. Mean iron intake in deferasirox and desferrioxamine recipients was 0.39 and 0.41 mg/kg/day, respectively. 1
II. Myocardial Iron Levels
Myocardial iron levels are estimated by calculating the T2* phase using magnetic resonance imaging. T2* intervals are inversely related to iron concentrations, as follows:
> 20 msec is considered normal
8 - 20 msec indicates mild to moderate myocardial iron overload
< 8 msec indicates severe myocardial iron overload
in vitro and in vivo preclinical data indicate that deferasirox has the capacity to enter and chelate iron from myocardial cells. Preliminary clinical data report that myocardial iron levels were significantly reduced after deferasirox treatment in patients with transfusional chronic iron overload.
In 22 patients aged 9 - 50 years with transfusional chronic iron overload who were treated with deferasirox 10 - 30 mg/kg/day, baseline T2* was increased from a geometric mean of 18.0 msec to 23.1 msec (p = 0.013) at 1 year. In a subsequent analysis, after 2 years of deferasirox treatment in 15 patients, myocardial T2* improved from a geometric mean of 16.1 msec at baseline to 22.5 msec at study end (p = 0.005).1
III. Labile Plasma Iron [LPI]
LPI, which may lead to oxidative damage of tissues, can be transported intracellularly and lead to excessive storage of iron. Consequently, it can be used as an indicator of iron overload and changes in LPI levels can be used to assess effectiveness of chelation therapy.
In 14 patients with ß-thalassaemia treated with deferasirox for 52 weeks, mean baseline LPI levels were 0.99 and 0.12 µmol/L pre- and post-dose (p < 0.0001), and study end values were 0.27 and 0.8 µmol/L, respectively. 1
Pharmacokinetic Properties1
Pharmacokinetic data pertaining to the recommended deferasirox dosage of 20 mg/kg/day are presented in Table 5. Tmax was independent of dose. After a single dose of deferasirox 20 mg/kg, median tmax for the Fe - [deferasirox]2 complex was 2.5 hours; median tmax ranged from 2 to 5 hours after a single dose of 2.5 - 80 mg/kg.
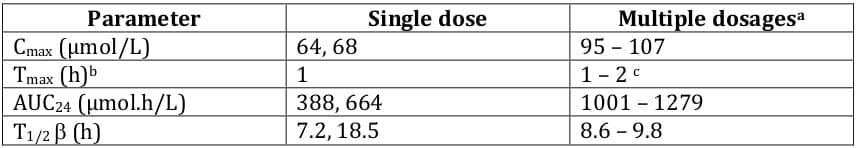
Mean Cmax and AUC of the Fe-[deferasirox]2 complex did not increase proportionally with dose, and substantial interindividual variability was observed. It is unclear whether increased systemic exposure to deferasirox correlates with increased iron chelation. 1
The pharmacokinetic properties of deferasirox (Tmax, Cmax, AUC and elimination t½) displayed dose linearity, indicating that use of a single daily dose was appropriate. 2
Orally administered deferasirox has an absolute bioavailability of 70%. The presence of food increased the bioavailability of deferasirox to a variable extent. To ensure appropriate systemic exposure, deferasirox should be taken 30 minutes before food. Deferasirox is highly protein bound (≈ 99%), almost exclusively to serum albumin.
The main metabolic pathway is hepatic glucuronidation, with excretion of glucuronide metabolites in bile. Deferasirox is mainly excreted in the faeces (83%), predominantly as the unchanged drug. A small proportion (6 - 8%) of unchanged deferasirox and metabolites is excreted in urine. 1
Impaired hepatic/renal function1
Although no pharmacokinetic data are available in patients with hepatic impairment, the kinetics of deferasirox were unchanged in patients with elevated ALT levels at baseline.
Deferasirox is minimally excreted by the renal route and renal impairment is not expected to affect the kinetics of deferasirox.
Potential drug interactions1
Aluminium
Although deferasirox has higher affinity for iron than for aluminium, the concomitant use of deferasirox and aluminium-containing antacids has not been studied and is not recommended.
Vitamin C
Vitamin C increases the availability of iron for chelation and patients with transfusional chronic iron overload are usually vitamin C deficient. The effects of concomitant administration of deferasirox and vitamin C have not been studied; however, vitamin C doses of ≤ 200 mg were permitted in clinical studies with no reported negative consequences.
The efficacy of oral deferasirox in patients with transfusional chronic iron overload has been evaluated in three randomised, open-label, multicentre trials comparing oral deferasirox with subcutaneous desferrioxamine 21, 22, 23 and two non-comparative multicentre trials. Trials were of 48 21, 24 or 52 22, 25, 23 weeks duration.
Three trials enrolled patients with ß-thalassaemia predominantly 21, 22, 24, and a fourth study enrolled patients with sickle cell disease. 23 Another trial had patients with myelodysplastic syndrome, Diamond-Blackfan syndrome and other rare anaemias. 25
One trial included only paediatric patients aged 2-17 years24, and one included only patients aged ≥ 18 years.21 The remaining trials included patients aged ≥ 2 years.22, 25, 23 In the latter trials, 49%22, 50%25 and 81%23 of patients were aged ≥ 16 years.
Additional uses under investigation include chronic haemodialysis-associated porphyria cutanea tarda and salvage therapy for rhinocerebral mucormycosis.
There have been no trials comparing deferasirox with deferiprone. Deferasirox has not been studied as part of combination therapy. The following section discusses the efficacy of deferasirox in comparison with desferrioxamine.
1. Phase II study in ß-thalassaemia(Haematologica 2006; 91: 873-80)
This study21 recruited 71 ß thalassaemia patients aged ≥ 18 years with transfusional haemosiderosis from four centers in Italy. 24 patients received deferasirox 10 mg/kg/day, whereas an additional 24 received deferasirox 20 mg/kg/day. 23 patients received desferrioxamine 40 mg/kg/day.
SQUID assessments at the end of the study showed average decreases in LIC of similar magnitude in the deferasirox 20 mg/kg/day and DFO groups (Figure 3).
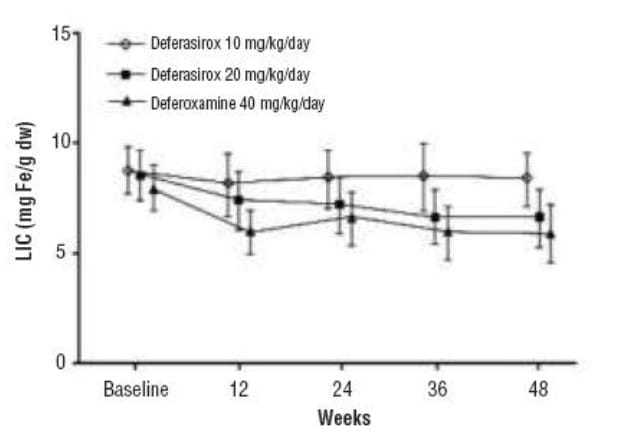
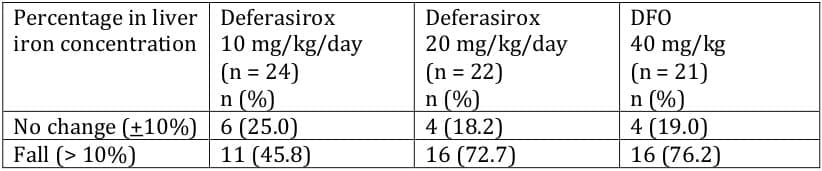
The number of responders, defined as patients exhibiting a > 10% fall in LIC by the end of the study, was equal in these two groups (Table 6). In contrast, deferasirox 10 mg/kg/day resulted in only a minimal fall in LIC.

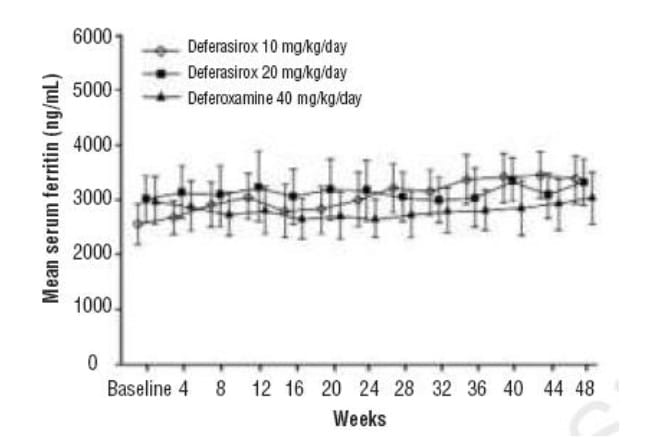
Over the study duration, mean serum ferritin levels remained stable in the deferasirox 20 mg/kg/day and DFO groups whereas there was a tendency for ferritin values to increase modestly over time in patients randomised to deferasirox 10 mg/kg/day (Figure 4). There were also no consistent changes in the levels of other markers of iron homeostasis (serum iron, transferrin and transferrin saturation).
Pharmacokinetic analysis showed that the plasma elimination half-life of deferasirox was 8-16 hours, supporting its once-daily administration.
In conclusion, this 48-week study demonstrated that deferasirox 20 mg/kg given orally once daily is efficacious in reducing LIC as DFO 40 mg/kg given by sc infusions for 5 days each week.
2. Phase 3 study in ß-thalassaemia (Blood 2006; 107: 3455-62)
This multinational phase 3 randomised trial 22 comparing deferasirox to desferrioxamine over 1 year was initiated in paediatric (≥ 2 years) and adult patients with ß-thalassaemia receiving regular blood transfusions. ß-thalassaemia was selected as the model disease for demonstration of efficacy across the range of patients at risk of iron overload.
Patients were randomised and received treatment with deferasirox (n = 296) or desferrioxamine (n = 290), with dosing according to baseline liver iron concentration (LIC). Approximately two thirds of each group was heavily iron overloaded as evidenced by an LIC value of 7 mg Fe/g dw or more at baseline (deferasirox 69%, desferrioxamine 68%).
Most patients completed 1 year of therapy on this study: 541 (92.3%) of 586 underwent both baseline and 1-year LIC assessments. Discontinuations were relatively similar in the groups receiving deferasirox (n = 17) and desferrioxamine (n = 12).
Changes in liver iron concentration
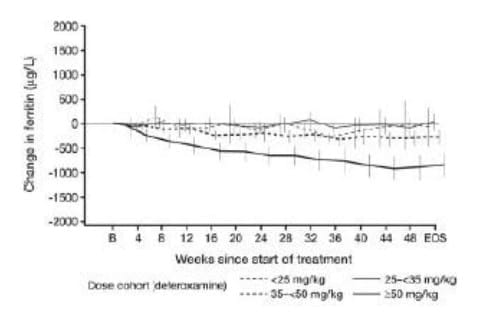
Deferasirox produced a dose-dependent reduction in absolute LIC (Figure 5).

About 60% of the patient population on deferasirox who completed end-of-study LIC evaluations had received daily doses of 20 or 30 mg/kg deferasirox for LIC of 7 mg Fe/g dw or higher at baseline. In this group, a statistically significant reduction in LIC was observed after 1 year (-5.3 ± 8.0 mg Fe/g dw, mean ± SD, p < 0.001). This reduction was similar to that effected by desferrioxamine (-4.3 ± 5.8 mg Fe/g dw, p = 0.367). However, efficacy was lower in patients who received 5-10 mg/kg/day deferasirox, indicating that this was not an effective dose.
Changes in serum ferritin
Effects on mean serum ferritin levels over time that paralleled the changes in LIC were observed in both deferasirox and desferrioxamine-treated patients (Table 6). Again, the 5-and 10-mg/kg/day dosages were less effective
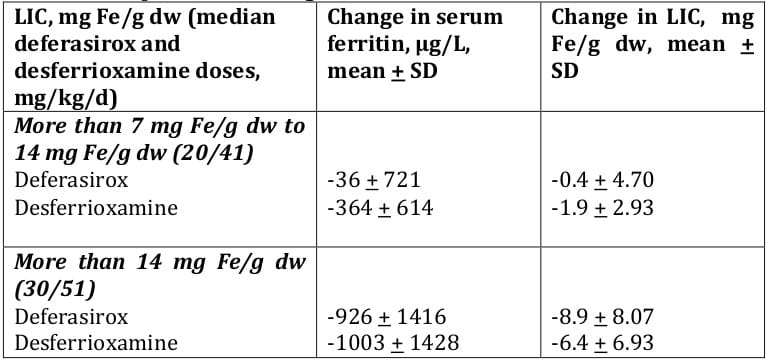
With monthly monitoring of serum ferritin values the response to different deferasirox doses could be distinguished after 12 weeks on study (Figure 5). Deferasirox doses of 20 mg/kg led to stable ferritin, and 30 mg/kg led to reduced ferritin values. Doses of 5 and 10 mg/kg were less effective.
Deferasirox
Changes in net body iron balance
Average iron intake per kilogram of body weight during the 1-year period of the study was similar in patients receiving deferasirox and desferrioxamine.
The ratios of iron excretion : iron intake indicate net iron excretion (values that are less than 1 indicate iron intake in excess of excretion, and values greater than 1 indicate iron excretion).
Table 7 depicts the net iron excretion.

Conclusion
Changes in serum ferritin, LIC levels, and net body iron balance over a 1-year interval were consistent. For both deferasirox and desferrioxamine, these 3 parameters corresponded well with one another.
In addition, a relationship between the dose of drug administered and the response observed was apparent for deferasirox.
- For patients receiving 5 mg/kg deferasirox and the majority of patients receiving 10 mg/kg deferasirox, all 3 parameters indicated an increase in iron burden.
- For those receiving 20 mg/kg deferasirox, they indicated that iron burden was essentially unchanged.
- For those receiving 30 mg/kg deferasirox, they indicated that iron burden was reduced.
3. Efficacy in sickle cell disease (Br J Haematol 2006; 136: 501-8)
In sickle cell disease, progressive iron loading and subsequent tissue injury appear similar to other transfused populations. Guidelines from consensus conferences have therefore recommended daily treatment with desferrioxamine in iron-overloaded patients.
Patients with sickle cell disease ≥ 2 years of age and with iron overload from repeated blood transfusions were enrolled in this trial.23 The serum ferritin level for entry into the screening period of this study was ≥ 1000 µg/l. A total of 195 adult and paediatric patients received deferasirox (n = 132) or desferrioxamine (n = 63). The majority of patients randomised to receive deferasirox were dosed at 10-30 mg/kg according to baseline LIC. The main efficacy endpoint was the change in LIC from baseline assessed at 52 weeks.
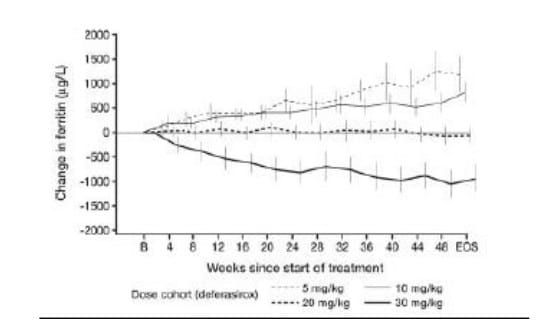
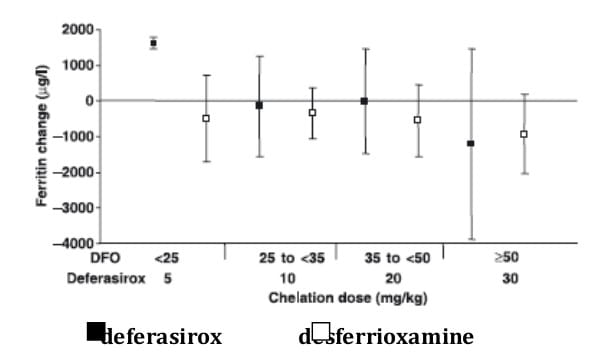
Administration of deferasirox resulted in a statistically significant reduction in LIC for the overall population, adjusted for transfusion category, of -3.0 ± 6.2 mg Fe/d dw (p < 0.001). This was similar to the reduction observed with desferrioxamine for the overall population adjusted for transfusion category, of -2.8 ± 10.4 Fe/g dw (p = 0.022) statistically significant reductions in LIC at the level of p < 0.05 were observed with treatment at deferasirox doses of 10-30 mg/kg and at a desferrioxamine dose of 35 to < 50 mg/kg (Figure 6). Similar reductions in LIC occurred across all age groups of patients enrolled into the trial.
Comparing patients who were categorised as only receiving simple transfusions to those only receiving exchange transfusions, the reductions in LIC observed with deferasirox were -1.6 ± 5.78 mg Fe/g dw (n = 62) and -6.6 ± 5.60 mg Fe/g dw (n = 22) respectively. The corresponding reductions in patients receiving desferrioxamine were -1.4 ± 3.12 mg Fe/g dw (n = 25) and -1.4 ± 3.90 (n = 10) respectively.
Changes in serum ferritin between baseline and end-of-study were dose dependent (Figure 7) and generally paralleled the changes in LIC observed.
Tolerability data for deferasirox were obtained from five trials of 48-52 weeks duration in paediatric and/or adult patients, with tolerability being the primary endpoint in three of these studies. Additional data are available from an ongoing extension study, in which patients had received deferasirox for a median of 91 weeks. 26
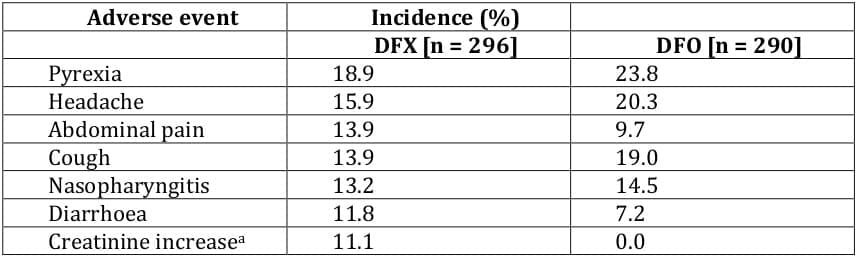
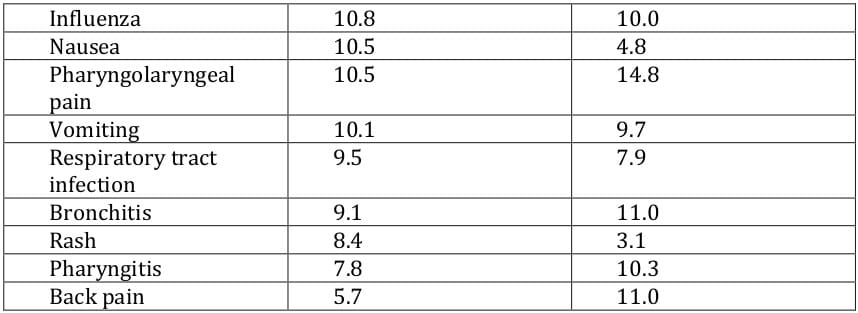
In clinical trials, deferasirox had a defined tolerability profile that was clinically manageable with regular patient monitoring, and the most common adverse events were usually mild and/or transient in nature and resolved spontaneously. 21, 22, 24, 25, 23 The most commonly occurring adverse events considered related to deferasirox included transient gastrointestinal adverse events (nausea, vomiting, abdominal pain, constipation and diarrhoea) and skin rash. 21, 22, 24, 25, 23 Adverse events in deferasirox recipients that required dosage adjustments or interruptions included rash, gastrointestinal disorders, infections and elevated serum creatinine and transaminase levels. 1 Dose-related adverse events with deferasirox included gastrointestinal symptoms, serum creatinine increase and skin rash. 1, 27 Withdrawals due to adverse events occurred in 2.4 - 8.9% of deferasirox and 0.3 - 8.7% of desferrioxamine recipients; 21, 25, 23, 26, 28, 29 these adverse events included arrhythmia, rash, increased ALT, diarrhoea, proteinuria and glycosuria. Serious adverse events, not considered related to the study drug, were reported in 46% of deferasirox recipients and in 43% of desferrioxamine recipients among patients aged ≥ 2 years, 23 and in 10% of paediatric patients receiving deferasirox. 24
The adverse events occurring most commonly in the phase III trial 22 are presented in Table 8. In terms of adverse events considered related to deferasirox, transient gastrointestinal adverse events occurred in 15% of patients and skin rash occurred in 11% of patients. 22 One deferasirox and three desferrioxamine recipients died during the study, although none of the deaths were considered to be related to the study drug. 22
The most common adverse events reported in phase II trials 21, 24, 25, 23 were generally consistent with those seen in the phase III trial. 22 In the study conducted solely in paediatric patients, the most commonly occurring adverse events were infections (mostly rhinitis and pharyngitis) and gastrointestinal adverse events (mostly vomiting and diarrhoea). 24
In the extension study, the most commonly occurring drug-related adverse events (nausea, diarrhoea, [upper] abdominal pain, vomiting and rash; incidence of 4.3 - 9.6%) were transient and mostly of mild severity. 26
Cardiac adverse events were reported in 5.1% of patients treated with deferasirox and 6.9% of desferrioxamine recipients, and serious cardiac adverse events were reported in ≤ 1% of patients with either drug. 22 No cardiac adverse events were attributable to deferasirox treatment. Prolongation of the QT interval was reported in two deferasirox recipients, but was resolved with treatment interruption and did not recur.
In studies involving paediatric patients, 22, 24,23, 26 growth and development in all deferasirox recipients was considered normal.1
Renal and Hepatic Adverse Events
In clinical trials, some deferasirox recipients developed increased serum creatinine levels.21, 22, 24, 25, 23 These increases were generally mild,22, 24, 25, 23 non-progressive,24, 25, 23 dose dependent22, 25 and often transient.22, 24 In one trial,23 36% of deferasirox and 22% of desferrioxamine recipients experienced mild and stable increases in serum creatinine; in another study, 22 38% of deferasirox and 14% of desferrioxamine recipients experienced mild, dose-dependent increases in serum creatinine. Serum creatinine levels generally remained within the normal range despite these increases;22, 24, 25, increases of > 33% from baseline were seen in 40% of deferasirox recipients. 25 Dose reductions because of increased serum creatinine levels occurred in 13% of patients in the pivotal phase III trial.22
In the extension study,26 increases in serum creatinine levels were more frequent in patients with the most rapid and greatest reduction in iron burden; most cases resolved spontaneously and the remainder were managed with dose reduction or interruption.
In terms of proteinuria, a urinary protein: creatinine ratio of ≥ 2 was reported in about 30% 23 and > 50% of deferasirox recipients in two clinical trials, and a urinary protein : creatinine ratio of > 1 was reported in 1% of deferasirox recipients in the long-term extension study. 26 No cases of (severe 25) renal insufficiency or renal failure were reported in trials.25, 26Postmarketing cases of renal failure have been reported in deferasirox recipients, with some fatalities; most of these patients had comorbidities, including advanced haematological disorders.30
Most patients receiving deferasirox had normal levels of serum ALT, although increased levels were reported in some patients.21, 22 Clinically significant increase in ALT (more thanfive times the ULN on two consecutive measurements) occurred in 3.8% of deferasirox recipients and in none of the desferrioxamine recipients.23 In a noncomparative trial in paediatric patients, increases requiring treatment interruption occurred in 12.5% of patients with deferasirox treatment.24
Other Adverse Events
In clinical trials in patients with ß-thalassaemia, no deferasirox-related agranulocytosis, or clinically significant (drug-related 24) neutropenia or thrombocytopenia, was reported. 21, 22, 24, 25 In addition, in patients with rare chronic anaemias, no worsening of myelosuppression was reported. 25 Postmarketing cases of deferasirox recipients developing cytopenias (including neutropenia and thrombocytopenia) have been reported; some of these patients have died. 30 It should be noted that most of these patients had pre-existing haematological disorders often associated with bone marrow failure. 30
Auditory or ophthalmological adverse events were reported infrequently in deferasirox recipients in some, 22, 26 but not all, 21, 24 clinical trials. For example, drug-related deafness, neurosensory deafness or hypoacusis was reported in 0.3% and 1.7% of patients treated with deferasirox or desferrioxamine, respectively. 22 Drug-related lens opacity or cataracts were reported in 0.3% of deferasirox recipients. 22
How should patients receiving Desirox be monitored?
Patients prescribed Desirox should be monitored as follows:
- Serum creatinine should be monitored, particularly in patients who are at increased risk of complications, have pre-existing renal conditions, are elderly, have co- morbid conditions, or are receiving medicinal products that depress renal function. Dosage reduction and/or temporary discontinuation may be required in patients who develop elevated serum creatinine.
- Liver function should be monitored regularly and treatment with deferasirox should be modified or interrupted if there are unexplained, persistent or progressive increases in serum transaminase levels.
- Patients should also have regular monitoring of blood counts and treatment should should be interrupted in patients who develop unexplained cytopenia.
MONITORING GUIDELINES

Deferasirox is an oral, once-daily iron chelator for the treatment of transfusional chronic iron overload. In the European Union, deferasirox is indicated in patients with ß thalassaemia major aged ≥ 6 years and, in the US, in all transfusional chronic iron overload patients aged ≥ 2 years.
Dosage
Prior to starting therapy, obtain baseline serum ferritin and iron levels. The risk for toxicity may be increased when deferasirox is given to patients with low iron burden or with serum ferritin levels that are only slightly elevated [see Dose Modifications].
The recommended initial daily dose of DESIROX is 20 mg/kg body weight.
Take DESIROX once daily on an empty stomach at least 30 minutes before food, preferably at the same time each day. Do not chew tablets or swallow them whole. Do not take DESIROX with aluminum-containing antacid products. Calculate doses (mg/kg per day) to the nearest whole tablet. Completely disperse tablets by stirring in water, orange juice, or apple juice until a fine suspension is obtained. Disperse doses of <1 g in 3.5 ounces of liquid and doses of ≥1 g in 7.0 ounces of liquid. After swallowing the suspension, resuspend any residue in a small volume of liquid and swallow.
Individualize the decision to remove accumulated iron based on anticipated clinical benefit and risks of DESIROX therapy. In patients who are in need of iron chelation therapy, it is recommended that therapy with DESIROX (deferasirox) be started when a patient has evidence of chronic iron overload, such as the transfusion of approximately 100 mL/kg of packed red blood cells (approximately 20 units for a 40-kg patient) and a serum ferritin consistently >1000 mcg/L.
Dose Modifications
DESIROX may require dose adjustment, interruption or cessation of the therapy due to toxicity or any of the following
Based on Serum Ferritin
After commencing initial therapy, monitor serum ferritin every month and adjust the dose of DESIROX if necessary every 3-6 months based on serum ferritin trends. Make dose adjustments in steps of 5 or 10 mg/kg and tailor adjustments to the individual patient's response and therapeutic goals (maintenance or reduction of body iron burden). In patients not adequately controlled with doses of 30 mg/kg (e.g., serum ferritin levels persistently above 2500 mcg/L and not showing a decreasing trend over time), doses of up to 40 mg/kg may be considered. Doses above 40 mg/kg are not recommended.
If the serum ferritin falls consistently below 500 mcg/L, consider temporarily interrupting therapy with DESIROX.
Based on Serum Creatinine
For adults, reduce the daily dose of DESIROX by 10 mg/kg if a rise in serum creatinine to >33% above the average of the pretreatment measurements is seen at 2 consecutive visits, and cannot be attributed to other causes. For pediatric patients, reduce the dose by 10 mg/kg if serum creatinine levels rise above the age-appropriate upper limit of normal at 2 consecutive visits.
Concomitant UGT inducers or Cholestyramine
Concomitant use of UGT inducers or cholestyramine decreases deferasirox systemic exposure (AUC). Avoid the concomitant use of cholestyramine or potent UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir) with DESIROX. If you must co-administer these agents together, consider increasing the initial dose of DESIROX to 30 mg/kg, and monitor serum ferritin levels and clinical responses for further dose modification.
Hepatic Impairment
Avoid the use of DESIROX in patients with severe (Child-Pugh C) hepatic impairment. Reduce the starting dose by 50% in patients with moderate (Child-Pugh B) hepatic impairment. Closely monitor patients with mild (Child-Pugh A) or moderate (Child- Pugh B) hepatic impairment for efficacy and adverse reactions that may require dose titration.
The benefits of iron chelation therapy in the treatment of patients with transfusional chronic iron overload are undisputed. Iron chelation therapy is recommended for patients requiring frequent blood transfusions, including those with thalassaemia, sickle cell disease or myelodysplastic syndrome.
Subcutaneous desferrioxamine was the first available chelator in the 1960s. However, its cumbersome mode of administration and cost precluded its widespread use.
The availability of the world's first oral iron chelator, Kelfer by Cipla ushered in a new era in the management of iron overload and helped extend the benefits of iron chelation therapy to thousands of children globally. In India, Kelfer is the most widely used iron chelator.
Deferasirox is a new, once-daily oral iron chelator available as dispersible tablets. With the weight of evidence available with clinical follow-up for over 2.6 years in more than 1,000 patients, the drug appears effective at stabilizing and reducing body iron and is a convenient and generally well-tolerated once-daily oral treatment.8 Oral deferasirox at a once-daily dose of 20 mg/kg led to maintenance of LIC, neutral iron balance, and stable serum ferritin levels. Once-daily doses of 30 mg/kg led to a significant reduction in LIC, negative iron balance, and decreased serum ferritin levels. 22 When combined with appropriate laboratory monitoring, the availability of deferasirox as a once-daily, oral option for safe and effective chelation therapy has the potential to prevent complications of iron overload. 23
Myocardial iron toxicity accounts for the majority of morbidity and mortality in patients with ß-thalassaemia. Deferasirox has shown efficacy in improving T2* in these patients, and preclinical data from animals suggest that deferasirox and deferiprone have similar efficacy in removing myocardial iron.1
The pharmacokinetic profile of deferasirox supports a convenient, once-daily, oral administration schedule, which offers the potential to improve compliance.21
In clinical studies of patients with a range of anaemic conditions, treatment with deferasirox was associated with a clinically manageable tolerability profile with regular patient monitoring.1
In summary, deferasirox represents an important option for the treatment of chronic iron overload due to blood transfusions.
Deferasirox Tablets
DESIROX
|
DESIROX may cause:
- renal impairment, including failure
- hepatic impairment, including failure
- gastrointestinal hemorrhage
In some reported cases, these reactions were fatal. These reactions were more frequently observed in patients with advanced age, high risk myelodysplastic syndromes (MDS), underlying renal or hepatic impairment or low platelet counts (<50 x 109/L) [see CONTRAINDICATIONS, WARNINGS AND PRECAUTIONS].
DESIROX therapy requires close patient monitoring, including measurement of:
- serum creatinine and/or creatinine clearance prior to initiation of therapy and monthly thereafter; in patients with underlying renal impairment or risk factors for renal impairment, monitor creatinine and/or creatinine clearance weekly for the first month, then monthly thereafter.
- serum transaminases and bilirubin prior to initiation of therapy, every two weeks during the first month and monthly thereafter.
|
DESIROX-500
Each tablet for oral suspension contains Deferasirox 500 mg
DESIROX-250
Each tablet for oral suspension contains deferasirox 250 mg
Tablets for oral suspension
Mechanism of Action
Deferasirox is an orally active chelator that is selective for iron (as Fe3+). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Although deferasirox has very low affinity for zinc and copper there are variable decreases in the serum concentration of these trace metals after the administration of deferasirox. The clinical significance of these decreases is uncertain.
Pharmacodynamics
Pharmacodynamic effects tested in an iron balance metabolic study showed that deferasirox (10, 20 and 40 mg/kg per day) was able to induce a mean net iron excretion (0.119, 0.329 and 0.445 mg Fe/kg body weight per day, respectively) within the clinically relevant range (0.1-0.5 mg/kg per day). Iron excretion was predominantly fecal.
Pharmacokinetics
Absorption
Deferasirox is absorbed following oral administration with median times to maximum plasma concentration (tmax) of about 1.5 to 4 hours. The Cmax and AUC of deferasirox increase approximately linearly with dose after both single administration and under steady-state conditions. Exposure to deferasirox increased by an accumulation factor of 1.3 to 2.3 after multiple doses. The absolute bioavailability (AUC) of deferasirox tablets for oral suspension is 70% compared to an intravenous dose. The bioavailability (AUC) of deferasirox was variably increased when taken with a meal.
Distribution
Deferasirox is highly (~99%) protein bound almost exclusively to serum albumin. The percentage of deferasirox confined to the blood cells was 5% in humans. The volume of distribution at steady state (Vss) of deferasirox is 14.37 ± 2.69 L in adults.
Metabolism
Glucuronidation is the main metabolic pathway for deferasirox, with subsequent biliary excretion. Deconjugation of glucuronidates in the intestine and subsequent reabsorption (enterohepatic recycling) is likely to occur. Deferasirox is mainly glucuronidated by UGT1A1 and to a lesser extent UGT1A3. CYP450-catalyzed (oxidative) metabolism of deferasirox appears to be minor in humans (about 8%). Deconjugation of glucuronide metabolites in the intestine and subsequent reabsorption (enterohepatic recycling) was confirmed in a healthy volunteer study in which the administration of cholestyramine 12 g twice daily (strongly binds to deferasirox and its conjugates) 4 and 10 hours after a single dose of deferasirox resulted in a 45% decrease in deferasirox exposure (AUC) by interfering with the enterohepatic recycling of deferasirox
Excretion
Deferasirox and metabolites are primarily (84% of the dose) excreted in the feces. Renal excretion of deferasirox and metabolites is minimal (8% of the administered dose). The mean elimination half-life (t1/2) ranged from 8 to 16 hours following oral administration.
Special Populations
Pediatric
Following oral administration of single or multiple doses, systemic exposure of adolescents and children to deferasirox was less than in adult patients. In children < 6 years of age, systemic exposure was about 50% lower than in adults.
Geriatric
The pharmacokinetics of deferasirox have not been studied in geriatric patients (65 years of age or older).
Gender
Females have a moderately lower apparent clearance (by 17.5%) for deferasirox
compared to males.
Renal Insufficiency
Deferasirox is minimally (8%) excreted via the kidney.
Hepatic Insufficiency
Deferasirox is principally excreted by glucuronidation and is minimally (8%) metabolized by oxidative cytochrome P450 enzymes. Deferasirox treatment has been initiated in patients with baseline liver transaminase levels up to 5 times the upper limit of the normal range. The pharmacokinetics of deferasirox were not influenced by such transaminase levels.
QT Prolongation
The effect of 20 and 40 mg/kg per day of deferasirox on the QT interval was evaluated in a single-dose, double-blind, randomized, placebo- and active-controlled (moxifloxacin 400 mg), parallel group study in 182 healthy male and female volunteers age 18-65 years. No evidence of prolongation of the QTc interval was observed in this study.
DESIROX (deferasirox) is indicated for the treatment of chronic iron overload due to blood transfusions (transfusional hemosiderosis) in patients 2 years of age and older. In these patients, deferasirox has been shown to reduce liver iron concentration and serum ferritin levels. Clinical trials to demonstrate increased survival or to confirm clinical benefit have not been completed.
Individualize the decision to initiate deferasirox therapy based on consideration of the anticipated clinical benefit and risks of the therapy, taking into consideration factors such as the life expectancy and comorbidities of the patient [see WARNINGS AND PRECAUTIONS AND CONTRAINDICATIONS].
The safety and efficacy of deferasirox when administered with other iron chelation therapy have not been established.
Dosing Information
Prior to starting therapy, obtain baseline serum ferritin and iron levels. The risk for toxicity may be increased when deferasirox is given to patients with low iron burden or with serum ferritin levels that are only slightly elevated [see Dose Modifications].
The recommended initial daily dose of DESIROX is 20 mg/kg body weight.
Take DESIROX once daily on an empty stomach at least 30 minutes before food, preferably at the same time each day. Do not chew tablets or swallow them whole. Do not take DESIROX with aluminum-containing antacid products. Calculate doses (mg/kg per day) to the nearest whole tablet. Completely disperse tablets by stirring in water, orange juice, or apple juice until a fine suspension is obtained. Disperse doses of <1 g in 3.5 ounces of liquid and doses of ≥1 g in 7.0 ounces of liquid. After swallowing the suspension, resuspend any residue in a small volume of liquid and swallow.
Individualize the decision to remove accumulated iron based on anticipated clinical benefit and risks of DESIROX therapy. In patients who are in need of iron chelation therapy, it is recommended that therapy with DESIROX (deferasirox) be started when a patient has evidence of chronic iron overload, such as the transfusion of approximately 100 mL/kg of packed red blood cells (approximately 20 units for a 40-kg patient) and a serum ferritin consistently >1000 mcg/L.
Dose Modifications
DESIROX may require dose adjustment, interruption or cessation of the therapy due to toxicity or any of the following [see WARNINGS AND PRECAUTIONS, Geriatric Use]
Based on Serum Ferritin
After commencing initial therapy, monitor serum ferritin every month and adjust the dose of DESIROX if necessary every 3-6 months based on serum ferritin trends. Make dose adjustments in steps of 5 or 10 mg/kg and tailor adjustments to the individual patient’s response and therapeutic goals (maintenance or reduction of body iron burden). In patients not adequately controlled with doses of 30 mg/kg (e.g., serum ferritin levels persistently above 2500 mcg/L and not showing a decreasing trend over time), doses of up to 40 mg/kg may be considered. Doses above 40 mg/kg are not recommended.
If the serum ferritin falls consistently below 500 mcg/L, consider temporarily interrupting therapy with DESIROX.
Based on Serum Creatinine
For adults, reduce the daily dose of DESIROX by 10 mg/kg if a rise in serum creatinine to >33% above the average of the pretreatment measurements is seen at 2 consecutive visits, and cannot be attributed to other causes. For pediatric patients, reduce the dose by 10 mg/kg if serum creatinine levels rise above the age-appropriate upper limit of normal at 2 consecutive visits.
Concomitant UGT inducers or Cholestyramine
Concomitant use of UGT inducers or cholestyramine decreases deferasirox systemic exposure (AUC). Avoid the concomitant use of cholestyramine or potent UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir) with DESIROX. If you must co-administer these agents together, consider increasing the initial dose of DESIROX to 30 mg/kg, and monitor serum ferritin levels and clinical responses for further dose modification [see Drug Interactions].
DESIROX is contraindicated in patients with:
- creatinine clearance <40 mL/min or serum creatinine >2 times the age-appropriate upper limit of normal;
- poor performance status and high-risk myelodysplastic syndromes or advanced malignancies [see WARNINGS AND PRECAUTION];
- platelet counts <50 x 109/L;
- known hypersensitivity to deferasirox or any component of DESIROX.
Renal
Acute renal failure, fatal in some patients and requiring dialysis in others, has been reported following the postmarketing use of deferasirox. Most of the fatalities occurred in patients with multiple comorbidities and who were in advanced stages of their hematological disorders. Monitor serum creatinine and/or creatinine clearance in patients who: are at increased risk of complications, have preexisting renal conditions, are elderly, have comorbid conditions, or are receiving medicinal products that depress renal function. Closely monitor the renal function of patients with creatinine clearances between 40 and less than 60 mL/min, particularly in situations where patients have additional risk factors that may further impair renal function such as concomitant medications, dehydration, or severe infections.
Assess serum creatinine and/or creatinine clearance in duplicate before initiating therapy to establish a reliable pretreatment baseline, due to variations in measurements. Monitor serum creatinine and/or creatinine clearance monthly thereafter. In patients with additional renal risk factors (see above), monitor serum creatinine and/or creatinine clearance weekly during the first month after initiation or modification of therapy and monthly thereafter.
Consider dose reduction, interruption, or discontinuation for increases in serum creatinine. If there is a progressive increase in serum creatinine beyond the age-appropriate upper limit of normal, interrupt DESIROX use. Once the creatinine has returned to within the normal range, therapy with DESIROX may be reinitiated at a lower dose followed by gradual dose escalation, if the clinical benefit is expected to outweigh potential risks.
In the clinical studies, for increases of serum creatinine on 2 consecutive measures (>33% in patients >15 years of age or >33% and greater than the age-appropriate upper limit of normal in patients <15 years of age), the daily dose of deferasirox was reduced by 10 mg/kg. Patients with baseline serum creatinine above the upper limit of normal were excluded from clinical studies.
In the clinical studies, deferasirox-treated patients experienced dose-dependent increases in serum creatinine. These increases occurred at a greater frequency compared to deferoxamine-treated patients (38% vs. 14%, respectively, in Study 1 and 36% vs 22%, respectively, in Study 3). Most of the creatinine elevations remained within the normal range (see “Undesirable Effects”). There have also been reports of renal tubulopathy in patients treated with deferasirox. The majority of these patients were children and adolescents with ß-thalassemia and serum ferritin levels <1500 mcg/L.
Hepatic dysfunction and failure
In study 1, 4 patients discontinued deferasirox because of hepatic abnormalities (drug-induced hepatitis in 2 patients and increased serum transaminases in 2 additional patients). There have been postmarketing reports of hepatic failure, some with a fatal outcome, in patients treated with deferasirox. Most of these events occurred in patients greater than 55 years of age. Most reports of hepatic failure involved patients with significant co-morbidities, including liver cirrhosis and multi-organ failure. Serum transaminases and bilirubin should be monitored before the initiation of treatment, every 2 weeks during the first month and monthly thereafter. Consider dose modifications or interruption of treatment for severe or persistent elevations.
Gastrointestinal
Fatal GI hemorrhages, especially in elderly patients who had advanced hematologic malignancies and/or low platelet counts, have been reported. Non-fatal upper GI irritation, ulceration and hemorrhage have been reported in patients, including children and adolescents, receiving deferasirox (see “UNDESIRABLE EFFECTS”). Physicians and patients should remain alert for signs and symptoms of GI ulceration and hemorrhage during DESIROX therapy and promptly initiate additional evaluation and treatment if a serious GI adverse event is suspected. Use caution when administering DESIROX in combination with drugs that have ulcerogenic or hemorrhagic potential, such as non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, oral bisphosphonates, or anticoagulants.
Cytopenias
There have been postmarketing reports (both spontaneous and from clinical trials) of cytopenias, including agranulocytosis, neutropenia and thrombocytopenia, in patients treated with deferasirox. Some of these patients died. The relationship of these episodes to treatment with deferasirox is uncertain. Most of these patients had preexisting hematologic disorders that are frequently associated with bone marrow failure (See “Undesirable Effects”). Monitor blood counts regularly. Consider interrupting treatment with DESIROX in patients who develop unexplained cytopenia. Reintroduction of therapy with DESIROX may be considered, once the cause of the cytopenia has been elucidated.
Hypersensitivity
Serious hypersensitivity reactions (such as anaphylaxis and angioedema) have been reported in patients receiving deferasirox, with the onset of the reaction occurring in the majority of cases within the first month of treatment (see “Undesirable Effects”). If reactions are severe, discontinue DESIROX and institute appropriate medical intervention.
Rash
Rashes may occur during DESIROX (deferasirox) treatment. For rashes of mild to moderate severity, DESIROX may be continued without dose adjustment, since the rash often resolves spontaneously. In severe cases, DESIROX may be interrupted. Reintroduction at a lower dose with escalation may be considered in combination with a short period of oral steroid administration. Erythema multiforme has been reported during deferasirox treatment.
Co-morbidities
Clinical trials to demonstrate increased survival or to confirm clinical benefit have not been completed. Deferasirox has been shown to decrease serum ferritin and liver iron concentration in clinical trials. Consider the importance of these factors as well as individual patient factors and the prognosis associated with any underlying conditions before initiation of DESIROX therapy [see CONTRAINDICATIONS].
In postmarketing experience, there have been reports of serious adverse reactions, some with a fatal outcome, in patients taking deferasirox therapy, predominantly when the drug was administered to patients with advanced age, complications from underlying conditions or very advanced disease. Most of these deaths occurred within six months of deferasirox initiation and generally involved worsening of the underlying condition. The reports do not rule out the possibility that deferasirox may have contributed to the deaths.
Special Senses
Auditory disturbances (high frequency hearing loss, decreased hearing), and ocular disturbances (lens opacities, cataracts, elevations in intraocular pressure, and retinal disorders) have been reported at a frequency of <1% with deferasirox therapy in the clinical studies. Auditory and ophthalmic testing (including slit lamp examinations and dilated fundoscopy) are recommended before starting deferasirox treatment and thereafter at regular intervals (every 12 months). If disturbances are noted, dose reduction or interruption should be considered.
Laboratory tests
Measure serum ferritin monthly to assess response to therapy and to evaluate for the possibility of overchelation of iron. If the serum ferritin falls consistently below 500 mcg/L, consider temporarily interrupting therapy with DESIROX (see “DOSAGE AND ADMINISTRATION”).
In the clinical studies, the correlation coefficient between the serum ferritin and LIC was 0.63. Therefore, changes in serum ferritin levels may not always reliably reflect changes in LIC.
Perform laboratory monitoring of renal and hepatic function (see “Warnings and Precautions”).
Drug Interactions
The concomitant administration of deferasirox and aluminum-containing antacid preparations has not been formally studied. Although deferasirox has a lower affinity for aluminum than for iron, do not administer DESIROX with aluminum-containing antacid preparations.
Effect of Deferasirox on Drug Metabolizing Enzymes
Deferasirox inhibits human CYP3A4, CYP2C8, CYP1A2, CYP2A6, CYP2D6, and CYP2C19 in vitro. The clinical significance of deferasirox inhibition of, CYP2A6, CYP2D6, and CYP2C19 is unknown.
Interaction with Midazolam and Other Agents Metabolized by CYP3A4
In healthy volunteers, the concomitant administration of deferasirox and midazolam (a CYP3A4 probe substrate) resulted in a decrease of midazolam peak concentration by 23 % and exposure by 17%. In the clinical setting, this effect may be more pronounced. Therefore, due to a possible decrease in CYP3A4 substrate concentration and potential loss of effectiveness, use caution when deferasirox is administered with drugs metabolized by CYP3A4 (e.g., cyclosporine, simvastatin, hormonal contraceptive agents).
Interaction with Repaglinide and Other Agents Metabolized by CYP2C8
In a healthy volunteer study, the concomitant administration of deferasirox (30 mg/kg/day for 4 days) and the CYP2C8 probe substrate repaglinide (single dose of 0.5 mg) resulted in an increase in repaglinide systemic exposure (AUC) to 2.3-fold of control and an increase in Cmax of 62%. If deferasirox and repaglinide are used concomitantly, consider decreasing the dose of repaglinide and perform careful monitoring of blood glucose levels. Exercise caution when deferasirox and other CYP2C8 substrates like paclitaxel are co-administered.
Interaction with Theophylline and Other Agents Metabolized by CYP1A2
In a healthy volunteer study, the concomitant administration of deferasirox (repeated dose of 30 mg/kg/day) and the CYP1A2 substrate theophylline (single dose of 120 mg) resulted in an approximate doubling of the theophylline AUC and elimination half-life. The single dose Cmax was not affected, but an increase in theophylline Cmax is expected to occur with chronic dosing. This increase in plasma concentrations could lead to clinically significant theophylline induced CNS or other adverse reactions. Avoid the concomitant use of theophylline or other CYP1A2 substrates with a narrow therapeutic index with deferasirox. If you must co-administer theophylline with deferasirox, monitor theophylline concentrations and consider theophylline dose modification.
Use caution when deferasirox is administered with other drugs metabolized by CYP1A2 such as cyclobenzaprine, imipramine, haloperidol, fluvoxamine, mexiletine, olanzapine, tizanidine, zileuton, and zolmitripan.
Interaction with Agents Inducing UDP-glucuronosyltransferase (UGT) Metabolism
In a healthy volunteer study, the concomitant administration of deferasirox (single dose of 30 mg/kg) and the potent UDP-glucuronosyltransferase (UGT) inducer rifampicin (600 mg/day for 9 days) resulted in a decrease of deferasirox systemic exposure (AUC) by 44%. Therefore, the concomitant use of deferasirox with potent UGT inducers (e.g., rifampicin, phenytoin, phenobarbital, ritonavir) may result in a decrease in deferasirox efficacy.
Avoid the concomitant use of potent UGT inducers with deferasirox. If you must co-administer these agents together, consider increasing the initial dose of deferasirox to 30 mg/kg and monitor serum ferritin levels and clinical responses for further dose modification [see Dosage and Administration].
Interaction with Cholestyramine
The concomitant use of deferasirox with cholestyramine may result in a decrease in deferasirox efficacy. In healthy volunteers, the administration of cholesytramine after a single dose of deferasirox resulted in a 45% decrease in deferasirox exposure (AUC). Avoid the concomitant use of cholestyramine with deferasirox. If you must co- administer these agents together, consider increasing the initial dose of deferasirox to 30 mg/kg and monitor serum ferritin levels and clinical responses for further dose modification [see Dosage and Administration].
Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies with deferasirox in pregnant women. Administration of deferasirox to animals during pregnancy and lactation resulted in decreased offspring viability and an increase in renal anomalies in male offspring at exposures that were less than the recommended human exposure. DESIROX should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In embryofetal developmental studies, pregnant rats and rabbits received oral deferasirox during the period of organogenesis at doses up to (100 mg/kg/day in rats and 50 mg/kg/day in rabbits) 0.8 times the MRHD (Maximum Recommended Human Dose) on a mg/m2 basis. These doses resulted in maternal toxicity but no fetal harm was observed.
In a prenatal and postnatal developmental study, pregnant rats received oral deferasirox daily from organogenesis through lactation day 20 at doses (10, 30, and 90 mg/kg/day) 0.08, 0.2, and 0.7 times the MRHD on a mg/m2 basis. Maternal toxicity, loss of litters, and decreased offspring viability occurred at 0.7 times the MRHD on a mg/m2 basis, and increases in renal anomalies in male offspring occurred at 0.2 times the MRHD on a mg/m2 basis.
Lactation
It is not known whether deferasirox is excreted in human milk. Deferasirox and its metabolites were excreted in breast milk of rats following a 10 mg/kg dose (about 0.08 times the recommended human oral dose based on body surface area). Because many drugs are excreted in human milk, caution should be exercised when deferasirox is administered to a nursing woman.
Pediatric Use
Of the 700 patients who received deferasirox during clinical studies, 292 were pediatric patients 2 - <16 years of age with various congenital and acquired anemias, including 52 patients age 2 - <6 years, 121 patients age 6 - <12 years and 119 patients age 12 - <16 years. Seventy percent of these patients had beta thalassemia. Children between the ages of 2 - <6 years have a systemic exposure to deferasirox approximately 50% of that of adults. However, the safety and efficacy of deferasirox in pediatric patients was similar to that of adult patients, and younger pediatric patients responded similarly to older pediatric patients. The recommended starting dose and dosing modification are the same for children and adults (see “Indications” and “Dosage and Administration”).
Growth and development were within normal limits in children followed for up to 5 years in clinical trials.
Geriatric Use
Four hundred and thirty-one (431) patients ≥65 years of age have been studied in clinical trials of deferasirox. The majority of these patients had myelodysplastic syndrome (MDS) (n=393). In these trials, elderly patients experienced a higher frequency of adverse reactions than younger patients. Closely monitor elderly patients for early signs or symptoms of adverse reactions that may require a dose adjustment. Elderly patients are at increased risk for deferasirox toxicity due to the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
Renal Impairment
Deferasirox has not been studied in patients with renal impairment. [See WARNINGS AND PRECAUTIONS]
Hepatic Impairment
Deferasirox has not been studied in patients with hepatic impairment.
Clinical Trials Experience
The following adverse reactions are also discussed in other sections of the labeling:
Renal Failure [see WARNINGS AND PRECAUTIONS]. Hepatic Failure [see WARNINGS AND PRECAUTIONS]. Fatal and non-fatal Gastrointestinal Bleedings [see WARNINGS AND PRECAUTIONS]. Cytopenias [see WARNINGS AND PRECAUTIONS].
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 700 adult and pediatric patients were treated with deferasirox for 48 weeks in premarketing studies. These included 469 patients with β-thalassemia, 99 with rare anemias, and 132 with sickle cell disease. Of these patients, 45% were male, 70% were Caucasian and 292 patients were <16 years of age. In the sickle cell disease population, 89% of patients were Black. Median treatment duration among these sickle cell patients was 51 weeks. Of the 700 patients treated, 469 (403 β-thalassemia and 66 rare anemias) were entered into extensions of the original clinical protocols. In ongoing extension studies, median durations of treatment were 88-205 weeks.
Table 1 displays adverse reactions occurring in >5% of deferasirox-treated β-thalassemia patients (Study 1) and sickle cell disease patients (Study 3) with a suspected relationship to study drug. Abdominal pain, nausea, vomiting, diarrhea, and skin rashes and increase in serum creatinine were the most frequent adverse reactions reported with a suspected relationship to deferasirox. Gastrointestinal symptoms, increases in serum creatinine, and skin rash were dose related.
|
Preferred Term
|
Study 1 (ß-Thalassemia)
|
Study 3 (Sickle Cell Disease)
|
|
Deferasirox N=296
n (%)
|
Deferoxamine N=290
n (%)
|
Deferasirox N=132
n (%)
|
Deferoxamine N=63
n (%)
|
|
Abdominal Pain**
Diarrhoea
Creatinine Increased***
Nausea
Vomiting
Rash
|
63 (21.3)
35 (11.8)
33 (11.1)
31 (10.5)
30 (10.1)
25 (8.4)
|
41 (14.1)
21 (7.2)
0 (0)
14 (4.8)
28 (9.7)
9 (3.1)
|
37 (28.0)
26 (19.7)
9 (6.8)
30 (22.7)
28 (21.2)
14 (10.6)
|
9 (14.3)
3 (4.8)
0
7 (11.1)
10 (15.9)
3 (4.8)
|
*Adverse reaction frequencies are based on adverse events reported regardless of relationship to study drug.
** Includes ‘abdominal pain’, ‘abdominal pain lower’, and ‘abdominal pain upper’ which were reported as adverse events.
*** Includes ‘blood creatinine increased’ and ‘blood creatinine abnormal’ which were reported as adverse events. Also see Table 2.
In Study 1, a total of 113(38%) patients treated with deferasirox had increases in serum creatinine >33% above baseline on 2 separate occasions (Table 2) and 25(8%) patients required dose reductions. Increases in serum creatinine appeared to be dose related (see “Warnings and Precautions). In this study, 17(6%) patients developed elevations in SGPT/ALT levels >5 times the upper limit of normal at 2 consecutive visits. Of these, 2 patients had liver biopsy proven drug-induced hepatitis and both discontinued deferasirox therapy (see “Warnings and Precautions”). An additional 2 patients, who did not have elevations in SGPT/ALT >5 times the upper limit of normal, discontinued deferasirox because of increased SGPT/ALT. Increases in transaminases did not appear to be dose related. Adverse reactions that led to discontinuations included abnormal liver function tests (2 patients) and drug-induced hepatitis (2 patients), skin rash, glycosuria/proteinuria, Henoch Schönlein purpura, hyperactivity/insomnia, drug fever, and cataract (1 patient each).
In Study 3, a total of 48(36%) patients treated with deferasirox had increases in serum creatinine >33% above baseline on 2 separate occasions (Table 2) (see “Warnings and Precautions”). Of the patients who experienced creatinine increases in Study 3, 8 deferasirox-treated patients required dose reductions. In this study, 5 patients in the deferasirox group developed elevations in SGPT/ALT levels >5 times the upper limit of normal at 2 consecutive visits and 1 patient subsequently had deferasirox permanently discontinued. Four additional patients discontinued deferasirox due to adverse reactions with a suspected relationship to study drug, including diarrhea, pancreatitis associated with gallstones, atypical tuberculosis, and skin rash.
|
Laboratory Parameter
|
Study 1 (ß-Thalassemia)
|
Study 3 (Sickle Cell Disease)
|
|
Deferasirox
N = 296
n (%)
|
Deferoxamine N=290
n (%)
|
Deferasirox N=132
n (%)
|
Deferoxamine
N=290
n (%)
|
|
Serum Creatinine
|
|
Creatinine increase >33% and <ULN at 2 consecutive post-baseline visits
|
113 (38.2)
|
41 (14.1)
|
48 (36.4)
|
14 ( 22.2)
|
|
Creatinine increase >33% and >ULN at 2 consecutive post-baseline visits
|
7 (2.4)
|
1 (0.3)
|
3 (2.3)
|
2 (3.2)
|
|
SGPT/ALT
|
|
SGPT/ALT >5 x ULN at 2 post-baseline visits
|
25 (8.4)
|
7 (2.4)
|
2 (1.5)
|
0
|
|
SGPT/ALT >5 x ULN at 2 consecutive post-baseline visits
|
17 (5.7)
|
5 (1.7)
|
5 (3.8)
|
0
|
Proteinuria
In clinical studies, urine protein was measured monthly. Intermittent proteinuria (urine protein/creatinine ratio >0.6 mg/mg) occurred in 18.6% of deferasirox-treated patients compared to 7.2% of deferoxamine-treated patients in Study 1. Although no patients were discontinued from deferasirox in clinical studies up to 1 year due to proteinuria, monthly monitoring is recommended. The mechanism and clinical significance of the proteinuria are uncertain.
Other Adverse Reactions
In the population of more than 5,000 patients who have been treated with deferasirox during clinical trials, adverse reactions occurring in 0.1% to 1% of patients included gastritis, edema, sleep disorder, pigmentation disorder, dizziness, anxiety, maculopathy, cholelithiasis, pyrexia, fatigue, pharyngolaryngeal pain, early cataract, hearing loss, gastrointestinal hemorrhage, gastric ulcer (including multiple ulcers), duodenal ulcer, and renal tubulopathy (Fanconi’s syndrome). Adverse reactions occurring in 0.01% to 0.1% of patients included optic neuritis and esophagitis. Adverse reactions which most frequently led to dose interruption or dose adjustment during clinical trials were rash, gastrointestinal disorders, infections, increased serum creatinine, and increased serum transaminases.
The following adverse reactions have been spontaneously reported during postapproval use of deferasirox. Because these reactions are reported voluntarily from a population of uncertain size, in which patients may have received concomitant medication, it is not always possible to reliably estimate frequency or establish a causal relationship to drug exposure.
Skin and subcutaneous tissue disorders: leukocytoclastic vasculitis, urticaria.
Immune system disorders: hypersensitivity reactions (including anaphylaxis and angioedema).
Cases of overdose (2-3 times the prescribed dose for several weeks) have been reported. In one case, this resulted in hepatitis which resolved without long-term consequences after a dose interruption. Single doses up to 80 mg/kg/day in iron overloaded beta thalassemic patients have been tolerated with nausea and diarrhea noted. In healthy volunteers, single doses of up to 40 mg/kg/day were tolerated. There is no specific antidote for deferasirox. In case of overdose, induce vomiting and employ gastric lavage.
Advise patients to take DESIROX once daily on an empty stomach at least 30 minutes prior to food, preferably at the same time every day. Instruct patients to completely disperse the tablets in water, orange juice, or apple juice, and drink the resulting suspension immediately. After the suspension has been swallowed, resuspend any residue in a small volume of the liquid and swallow. Advise patients not to chew or swallow the tablets whole.
Advise patients not to chew tablets or swallow them whole.
Advise patients who experience diarrhea or vomiting to maintain adequate hydration.
Caution patients not to take aluminum-containing antacids and DESIROX simultaneously.
Because auditory and ocular disturbances have been reported with deferasirox, conduct auditory testing and ophthalmic testing before starting DESIROX treatment and thereafter at regular intervals (see “WARNINGS AND PRECAUTIONS”).
Caution patients experiencing dizziness to avoid driving or operating machinery (see “UNDESIRABLE EFFECTS”).
Caution patients about the potential for the development of GI ulcers or bleeding when taking DESIROX in combination with drugs that have ulcerogenic or hemorrhagic potential, such as NSAIDs, corticosteroids, oral bisphosphonates, or anticoagulants.
Caution patients about potential loss of effectiveness of drugs metabolized by CYP3A4 (e.g., cyclosporine, simvastatin, hormonal contraceptive agents) when DESIROX is administered with these drugs.
Caution patients about potential loss of effectiveness of Desirox when administered with drugs that are potent UGT inducers (e.g. rifampicin, phenytoin, phenobarbital, ritonavir). Based on serum ferritin levels and clinical response, consider increases in the dose of Desirox when concomitantly used with potent UGT inducers.
Perform careful monitoring of glucose levels when repaglinide is used concomitantly with Desirox. An interaction between Desirox and other CYP2C8 substrates like paclitaxel cannot be excluded.
Advise patients that blood tests will be performed because DESIROX may affect your kidneys, liver, or blood. The blood tests will be performed every month or more frequently if you are at increased risk of complications (e.g., pre-existing kidney condition, are elderly, have multiple medical conditions, or are taking medicine that affects your organs). There have been reports of severe kidney and liver problems, blood disorders, stomach bleeds and death in patients taking deferasirox.
Skin rashes may occur during DESIROX treatment and if severe treatment should be interrupted. Serious allergic reactions (which include swelling of the throat) have been reported in patients taking deferasirox, usually within the first month of treatment. If reactions are severe, advise patients to stop taking DESIROX and contact their doctor immediately.
Certain patients should not receive DESIROX. These include patients with creatinine clearance <40 mL/min or serum creatinine >2 times the age-appropriate upper limit of normal, patients with poor performance status and high-risk myelodysplastic syndromes or advanced malignancies, patients with platelet counts <50 x 109/L, and those with hypersensitivity to deferasirox or any component of DESIROX.
Desirox – 250 mg --------------Bottles of 30 tablets
Desirox – 500 mg --------------Bottles of 30 tablets
- Yang LPH, Keam SJ and Keating GM. Deferasirox: A review of its use in the management of transfusional chronic iron overload. Drugs 2007; 67: 2211-30
- Lindsey WT and Olin BR. Deferasirox for transfusion-related iron overload: A clinical review. Current Therapeutics 2007; 29: 2154 – 66
- Barton JC. Deferasirox. Current Opinion in Investigational Drugs 2005; 6: 327-35
- Fischer R. The oral chelator deferasirox - a new perspective for patients with iron overload. Haematologica 2006; 91: 865
- Deferasirox: A new iron chelator. The Medical Letter 2006; 48: 35 – 36
- Kontoghiorghes GJ. Deferasirox: uncertain future following renal failure fatalities, agranulocytosis and other toxicities. Expert Opinion Drug Safety 2007; 6: 235-39
- Dubey AP, Sudha S and Parakh A. Defarasirox: The new oral iron chelator. Indian Paediatrics 2007; 44: 603-7
- Porter JB. Deferasirox: An effective once-daily orally active iron chelator. Drugs of Today 2006; 42: 623 – 37
- Hershko C. Oral iron chelators: new opportunities and new dilemmas. Haematologica 2006; 91: 1307 – 12
- Neufeld EJ. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassaemia major: new data, new questions. Blood 2006; 107; 3436-41
- Van Orden HE and Hagemann TM. Deferasirox – An oral agent for chronic iron overload. Annals of Pharmacotherapy 2006; 40: 1110-7
- Shashaty G et al. Deferasirox for the treatment of chronic iron overload in transfusional hemosiderosis. Oncology 2006; 20: 1799
- Stumpf JL. Deferasirox. Am J Health-System Pharmacy 2007; 64: 606-16
- Hershko C et al. Objectives and mechanism of iron chelation therapy. Annals New York Academy of Sciences 2005; 1054: 124 – 35.
- Barton JC. Optimal management strategies for chronic iron overload. Drugs 2007; 67: 685-700.
- Piga A et al. Comparative effects of deferiprone and desferrioxamine on survival and cardiac disease in patients with thalassaemia major: a retrospective analysis. Haematologica 2003; 88: 489-96
- Anderson LJ et al. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in b-thalassaemia. Lancet 2002; 360: 516 – 20.
- Pennell DJ et al. Randomised controlled trial of deferiprone or desferrioxamine in b-thalassaemia major patients with asymptomatic myocardial siderosis. Blood 2006; 107: 3738-44
- Borgna-Pignatti C et al. Cardiac morbidity and mortality in desferrioxamine or deferiprone-treated patients with thalassaemia major. Blood 2006; 107: 3733-7
- Choudhry VP and Naithani R. Current status of iron overload and chelation with deferasirox. Indian Journal of Paediatrics 2007; 74: 759-64
- Piga A et al. Randomised phase II trial of deferasirox, a once-daily, orally administered iron chelator, in comparison to deferoxamine in thalassaemia patients with transfusional iron overload. Haematologica 2006; 91: 873-80
- Cappellini MD et al. A phase 3 study of deferasirox, a once-daily oral iron chelator, in patients with in b-thalassaemia. Blood 2006; 107: 3455-62
- Vichinsky E et al. A randomized comparison of deferasirox versus deferoxamine for the treatment of transfusional iron overload in sickle cell disease. British Journal of Haematology 2006; 136: 501-08
- Galanello R et al. Phase II clinical evaluation of deferasirox, a once-daily oral chelating agent, in paediatric patients with b-thalassaemia major. Haematologica 2006; 91:1343-51
- Porter J et al. A phase II study with ICL670, a once-daily oral iron chelator, in patients with various transfusion-dependent anemias and iron overload (abstract no 3193). Blood 2004; 104: 872a Plus poster presented at the 46th Annual Meeting and Exposition of the American Society of Haematology; 2004 Dec 4-7; San Diego (CA).
- Cappellini MD, Giardina P, Porter T et al. Long term safety and tolerability of the once daily oral iron chelator deferasirox in patients with transfusional iron overload (abstract no 1768). Blood 2006 Nov; 108(11 Pt 1): 501a. Plus poster presented at the 48th Annual Meeting and Exposition of the American Society of Haematology; 2006 Dec -12; Orlando (FL)
- Cappellini MD. Deferasirox is clinically effective and generally well tolerated, and demonstrates dose-responsiveness (letter).Blood 2006; 108: 775-6
- European Medicines Agency. European public assessment report (EPAR): scientific discussion http://www.emea.europa.eu/humandocs/PDFs/EPAR/exjade/H-670-en6.pdf Accessed 10th April 2008
- 29. Shashaty G. Food and Drug Administration Blood Products Advisory Committee: FDA briefing document (clinical review). http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4177B1_02_b.pdf Accessed 10th April 2008
- 30. Hohnekar JA. Important drug warning.
http://www.fda.gov/medwatch/safety/2007/Exjade_DHCPL_May2007.pdf Accessed 10th April 2008