Rioteph (Riociguat) Monograph



Chronic Thromboembolic Pulmonary Hypertension (CTEPH): Overview
Chronic thromboembolic pulmonary hypertension (CTEPH) is a progressive pulmonary vascular disorder. Classified as group 4 pulmonary hypertension (PH) by the joint task force of European Society of Cardiology (ESC)/European Respiratory Society (ERS)1.
1. Pulmonary arterial hypertension
1.1 Idiopathic
1.2 Heritable (BMPR2 mutation, Other mutations)
1.3 Drugs and toxins induced
1.4 Associated with:
1.4.1 Connective tissue disease
1.4.3 Portal hypertension
1.4.4 Congenital heart disease
1.4.5 Schistosomiasis
1’. Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis
1”. Persistent pulmonary hypertension of the newborn
2. Pulmonary hypertension due to left heart disease
2.1 Left ventricular systolic dysfunction
2.2 Left ventricular diastolic dysfunction
2.3 Valvular disease
2.4 Congenital /acquired left heart inflow/out flow tract obstruction and congenital cardiomyopathies
2.5 Congenital /acquired pulmonary veins stenosis
3. Pulmonary hypertension due to lung diseases and/or hypoxia
3.1 Chronic obstructive pulmonary disease
3.2 Interstitial lung disease
3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4 Sleep-disordered breathing
3.5 Alveolar hypoventilation disorders
3.6 Chronic exposure to high altitude
3.7 Developmental lung diseases
4. Chronic thromboembolic pulmonary hypertension and other pulmonary artery obstructions
4.1 Chronic thromboembolic pulmonary hypertension
4.2 Other pulmonary artery obstructions
4.2.1 Angiosarcoma
4.2.2 Other intravascular tumors
4.2.3 Arteritis
4.2.4 Congenital pulmonary arteries stenoses
4.2.5 Parasites (hydatidosis)
5. Pulmonary hypertension with unclear and/or multifactorial mechanisms
5.1 Haematological disorders
5.2 Systemic disorders
5.3 Metabolic disorders
5.4 Others
CTEPH is characterised by macroscopic thromboembolic lesions within proximal or distal pulmonary arteries and microscopic pulmonary vasculopathy. The clinical consequences of these pathological changes, which can include intimal thickening, vascular remodelling and plexiform lesions, are elevated pulmonary vascular pressures and increased pulmonary vascular resistance (PVR), raising the risk of right heart failure and death.2
CTEPH, if left untreated, have a poor prognosis with survival rates at 5 years of 30% for patients with mean pulmonary arterial pressure (mPAP) >40 mmHg, and 10% for patients with an mPAP >50 mm Hg.2
Burden of Disease
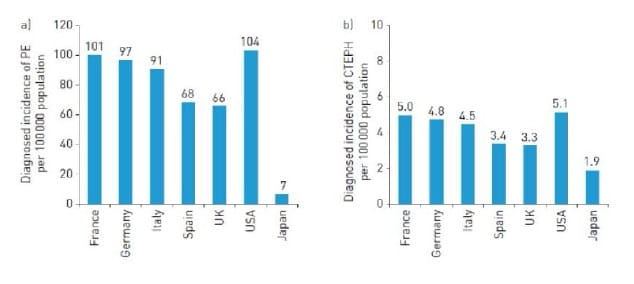
Epidemiological data on CTEPH is limited from across the world. A recent literature review analysed data from across US, Europe and Japan, and provided estimates of the incidence. A crude annual incidence of diagnosed pulmonary embolism and crude annual full (i.e. diagnosed and undiagnosed) incidence of CTEPH of 66–104 and 3–5 cases per 100000 population was reported from US and Europe.3 (Fig 1)
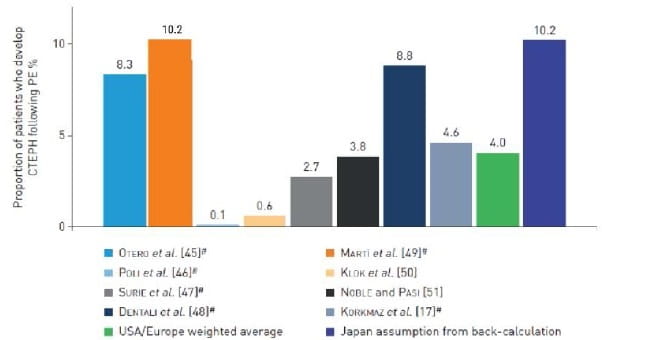
While, the incidence of CTEPH in patients following an episode of PE ranges from 0.1% to 9.1%, with a calculated weighted average of 4% (Fig 2).3
In 2013, over all 7–29% of CTEPH cases in Europe and the USA were diagnosed, and most patients were in New York Heart Association functional class III/IV at diagnosis. 3
The time taken for the development of CTEPH following episode of pulmonary embolism
CTEPH can develop several months or years after an acute PE (which may be silent), despite continuing anticoagulation, and in the absence of new symptoms or any new acute event 4, 5 (reviewed in Simmoneau et al 2017).
A widely regarded Italian study of 223 consecutive patients with acute PE found a cumulative CTEPH incidence of 3.8% after 2 years of index PE; but no additional CTEPH cases were identified during long-term follow-up of 10 years.6 (Fig 3)
Cumulative incidence of development of CTEPH after acute PE was found to be: 6
1% at 6 months
3.1% at 1 year
3.8% at 2 years
.jpg?updated=20230105110944)
A European registry in 2011 demonstrated that 74.8% of CTEPH patients presented with previous acute PE. 7
However, patients with CTEPH can present without a history of PE, but studies vary widely in their estimates of what proportion such patients represent. These patients range from 25% to 63%. 8-11
Therefore, the true incidence of CTEPH is likely to be underestimated by studies that only follow patients after a PE and highlights the fact that all patients with PH should be assessed for CTEPH even in the absence of prior PE. 7-11
Incidence in India
|
“PE is an under-recognised but likely common clinical problem in India and given that a significant proportion of patients with acute PE may progress to CTEPH even within one year after the index event, aggressive follow-up and appropriate management is needed to improve outcomes in this potentially fatal condition.” Indian J Chest Dis Allied Sci 2013; 55:205-207
|
There is no CTEPH registry in India. However, a recent observational screening study in India from Narayan Hrudayalaya hospital Bangalore, revealed a 52% incidence of acute PE of which 20 % of patients progressed to CTEPH in one-and-a half-years’ time. 12
Pathophysiology of CTEPH
The mechanism of pulmonary hypertension in CTEPH is multifactorial. Natural history of CTEPH is outlined in Fig 4.
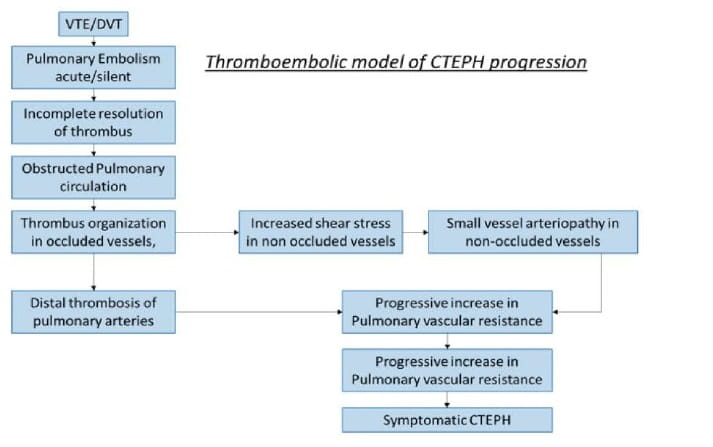
Though CTEPH usually begins with persistent obstruction of large and or middle sized pulmonary arteries by organised thrombi, recent insights have revealed that CTEPH involves not only persistent organized thrombi in proximal pulmonary arteries (main, lobar and segmental) but also in small vessels in the obstructed area which also contributes to hemodynamic compromise, functional impairment and disease progression.2
Risk Factors and Causes of CTEPH
Risk factors that predisposes patients to CTEPH include: 13,14
Pulmonary embolism: CTEPH is generally considered to be rare and late complication of one or more episodes of acute PE that fail to resolve despite more than 3 months of curative anti-coagulation. Large pulmonary emboli appear to carry a higher risk of progression to CTEPH, probably because the lytic machinery is unable to dissolve large embolus entirely. It can develop several months or years after an acute PE despite anti-coagulation treatment and in absence of new symptoms or new acute event. 5
- Haemostatic risk factors:
o Elevated levels of pro-coagulant proteins: Factor VIII and Von Willebrand factors, Plasminogen activator inhibitor-1 (PAI-1)
o Modification in the fibrinogen to lysis resistant variant
o Non-O blood group
- Associated medical conditions:
o Splenectomised patients
o Ventriculoarterial shunt surgeries in hydrocephalic patients
o Infected pacemaker
o Chronic inflammatory conditions: osteomyelitis, inflammatory bowel disease
o Antiphospholipid syndrome (autoimmune condition)
o Hypothyroidism and treatment with levothyroxine
o Cancer patients
Symptoms of CTEPH
Many people who develop CTEPH after PE go through a so-called “honeymoon period” when they do not have any symptoms. When symptoms develop, they are vague and non-specific in the early stages of the disease. These include:
- Feeling short of breath with activity
- Tiredness and fatigue
- Depressed mood
- Syncope
Symptoms of later-stage disease include fainting and signs of right heart failure, such as fluid retention (oedema) and blue-tinged fingers and toes (cyanosis). Some patients with CTEPH never have any early symptoms, therefore called ‘silent’.
Standard of Care and the Changing Landscape
Diagnosis of CTEPH is based on
CTEPH diagnosis is challenging, disease progression takes place quickly, so detecting CTEPH early is vital.
Diagnosis is based on 1
1. History of DVT or PE: Previously reported episode of pulmonary embolism or deep vein thrombosis.
2. Clinical findings (signs, symptoms)
Symptoms: Breathlessness with exercise, chest discomfort, fatigue, palpitations, depressed mood, light-headedness and even passing out with exercise. Rarely, coughing up blood can occur.
Signs: Dyspnea mainly with exercise or activity, fatigue, syncope during physical activity (passing out), hemoptysis (seen in some patients). Some patients have swelling of their legs.
3. Imaging studies:
i) Echocardiogram: To screen pulmonary hypertension and to evaluate effect on right ventricle. Parameters such as peak tricuspid regurgitation velocity (TRV), RV hypertrophy, RV over load, and hemodynamic changes are evaluated.
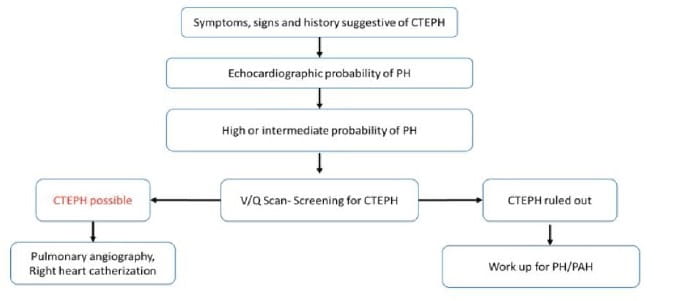
ii) V/Q scan: The V/Q scan is the screening method of choice and the first imaging modality for CTEPH, because of its sensitivity (96-97%) and specificity (90-95%). Its sensitivity is higher than CTPA. However, in some cases, a normal V/Q scan doesn’t necessarily mean the patient does not have CTEPH.
iii) Computed tomography (CT) pulmonary angiogram: Contrast CT angiography of pulmonary artery is helpful in determining whether there is evidence of surgically accessible CTEPH. It can delineate the typical angiogenic findings in CTEPH, such as complete obstruction, bands and webs and intimal abnormalities accurately and reliably.
iv) Right Heart catheterization - checks blood pressure and flow in the heart’s arteries and chambers.
The diagnostic algorithm recommended by ERS/ESC 2015 is described in figure 5.
4. Biochemical tests 15: A work up of biochemical or haematological blood tests of patients is generally performed. Increased levels of lupus anticoagulant/anti-cardiolipin antibodies and Factor VIII have been reported in a proportion of patients with CTEPH. N-terminal pro-brain natriuretic peptide (NT pro-BNP) is also commonly used biomarker for CTEPH. Normal N-terminal proBNP levels in conjunction with ECG characteristics have been proposed as “CTEPH rule-out criteria” following acute PE. Other tests, such as D-dimer, troponin, C-reactive protein, heart-type fatty acid-binding protein and recently pentraxin 3, have also been reported as predictors of outcome in CTEPH, although their diagnostic potential has not been systematically studied.
Treatment:
CTEPH is the only PH group which is potentially curable.
Pulmonary endarterectomy, a surgical procedure is the 1st line of treatment and was considered as the only effective and recommended treatment for patients with CTEPH. This provides major clinical and haemodynamic improvements and is associated with increased survival. However, this procedure is invasive and can pose too high a risk to some patients. Further, 24-37% of CTEPH patients are not eligible for PEA and 17-35% patients experience persistent and recurrent PH after PEA.2
Operability depends on the nature and location of the thrombi, and the extent of the underlying micro vasculopathy, and other co-morbidities.
Balloon pulmonary angioplasty (BPA) or percutaneous transluminal pulmonary angioplasty, is an emerging alternative management option for some patients, who may, for example, have lesions too distal to be treated by pulmonary endarterectomy.2
Overall, the treatment selection depends on size and the site of the emboli or fibrotic clots in the pulmonary vasculature (Fig 6).
- PEA is the treatment of choice in CTEPH if the clots are present in the proximal region of the pulmonary vasculature in operable patients.
- BPA can be performed when the clots are present in segmental and sub-segmental branches.
- In cases of microvasculopathy, in which small vessels are affected and in cases of persistent or recurrent cases, medical treatment is the only viable option.
.jpg?updated=20230105111439)
Thus, the four distinct clinical situations in patients with CTEPH where pulmonary arterial hypertension (PAH) targeted therapy may work, include:
1. Distal CTEPH not amenable to pulmonary endarterectomy.
2. Residual pulmonary hypertension after endarterectomy.
3. Non-operable CTEPH due to medical comorbidities or having surgery declined by the patient.
4. Bridging therapy to endarterectomy in those with hemodynamically severe disease
PAH-targeted therapy including prostanoids, phosphodiesterase type 5 inhibitors, endothelin receptor antagonists, have been considered for use in CTEPH. However, these studies demonstrated mixed results.
In this context, the soluble guanylate cyclase stimulator riociguat, is a promising introduction to medical management, targeting microvascular disease in patients with inoperable CTEPH, and those with persistent or recurrent CTEPH following surgical treatment. Its use is currently recommended in the ESC/ERS guidelines. 1
|
Development of riociguat is a milestone for patients with inoperable or recurrent/ persistent CTEPH. Riociguat was the first drug in its class approved for PAH and was the first FDA-approved medication approved for treating patients with WHO group 4 PH, also known as chronic thromboembolic pulmonary hypertension (CTEPH). |
Riociguat
Riociguat is the first and only approved drug for CTEPH for both inoperable and for persistent/or recurrent CTEPH.
Riociguat belongs to a new class of pharmacologic agents called soluble guanylate cyclase stimulator
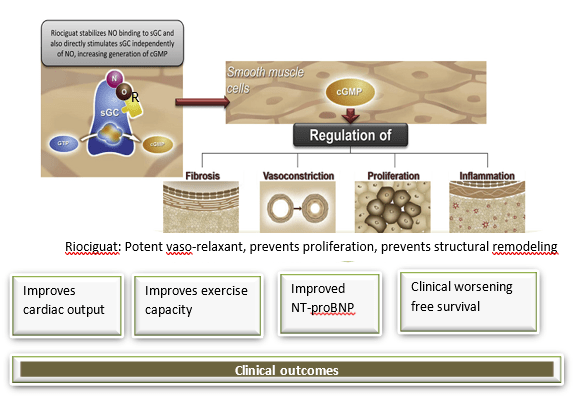
The Role of the NO-sGC-cGMP Signalling Pathway
Endothelium cell-derived NO acts on vascular smooth muscle cells in healthy subjects to induce vasodilatation by increasing production of the secondary messenger cyclic guanosine monophosphate (cGMP) via activation of soluble guanylate cyclase (sGC). 16
In healthy lungs: 16
1. NO is a key regulator of flow-induced vasodilatation in the lung (Fig 7). In the healthy lung, NO is produced by the vascular endothelium, airway, and alveolar epithelial cells and diffuses to the smooth muscle layer, where it binds and activates to soluble guanylate cyclase( sGC).
2. Activated sGC converts guanosine-5’-triphosphate to the intracellular second messenger cGMP.
3. Increase in the cGMP concentration can affect multiple pathways, including activation of protein kinase G.
4. Protein kinase G activation leads to a decrease in intracellular calcium, inhibition of the actin–myosin contractile system and vascular smooth muscle relaxation.
Thus in the healthy lung, NO plays a key role in the maintenance of a low vascular tone and a normal ventilation/ perfusion matching. Levels of NO are low in regions of low ventilation, resulting in vasoconstriction and high in regions of high ventilation, resulting in vasodilatation. This causes blood flow to be directed preferentially toward well-ventilated lung, ensuring efficient oxygenation.

In lungs of CTEPH patient
Traditional targeting of this pathway is to increase the amount of bioavailable cGMP in the smooth muscle of the pulmonary vasculature through use of PDE5 inhibitors such as sildenafil or tadalafil or, in some cases, gaseous NO. PDE5 inhibitors rely upon NO-sGC signalling in order to catalyze the synthesis of cGMP and thus confer its therapeutic effects. However, PH including CTEPH causes phenotypic alterations in NO synthesis there by resulting in inadequate response of PDE5i or NO gas treatment.
The NO signalling pathway is impaired in CTEPH and the reduction of NO could be due to multiple mechanisms 16,17 (Shown in Red arrows, Figure 8)
1. Down-regulation or uncoupling of endothelial nitric oxide synthase (eNOS) (denoted as step1 in Figure 8)
2. Upregulation of arginase activity and depletion of L-arginine (substrate for NO synthesis) (denoted as step 2, red arrow)
3. eNOS inhibition (red blocked arrow) due to increased circulating levels of dimethyl arginine (denoted as step 3)
4. Inactivation of NO by reactive oxygen species (denoted as step 4)
Fig 8: Dysregulation of nitric oxide pathway in CTEPH patients. Nitric oxide synthesis can be impaired through increased activity of arginase, decreased availability of L-arginine and eNOS, uncoupling of eNOS from co-factors and increased availability of eNOS inhibitors like ADMA. ADMA: asymmetric dimethylarginine; BH2: dihydrobiopterin; BH4: tetrahydrobiopterin; GMP: guanosine monophosphate; GTP: guanosine triphosphate; O2– : superoxide anion, sGC: soluble guanylate cyclase.
Riociguat: Mode of Action
Riociguat has dual mode of action: 16,17
a) It directly stimulates sGC in an NO-independent causing vasodilation
b) It is also capable of synergizing with NO, by increasing the sensitivity of sGC to NO over a wide range of concentrations and stabilises the NO-sGC interaction.
The NO independent action of riociguat distinguishes it from PDE-5 inhibitors, which require NO to function and may have limited efficacy in the presence of very low NO levels. 13
In vitro, riociguat had 12 times more potent vasorelaxant properties in nitrate-tolerant vessels compared with the NO donor glycerol trinitrate.17Fig 9: Summary of the mode of action and effects of riociguat. R: Riociguat, NO: Nitric oxide, GTP: Guanosine triphosphate, cGMP: cyclic guanosine monophosphate, sGC, soluble guanylate cyclase, NT-proBNP (N-terminal pro-brain natriuretic peptide).
In vitro, riociguat produces an approximately 73-fold increase in sGC activity independent of NO, whereas in the presence of NO, it produces approximately a 122-fold increase. 17
There were no reports of sensitization or tachyphylaxis with riociguat or active metabolite M1 ( ~ 1/3rd – 1/10th the potency of riociguat). 18
|
In vitro and preclinical studies have shown that riociguat have potent vasorelaxant properties, prevents cardiopulmonary remodeling, improves hemodynamics and gas exchange. CHEST 2017; 151: 468-480. |
Riociguat-Pharmacokinetics
The pharmacokinetics of riociguat has been evaluated in both healthy volunteers and in patients of CTEPH. 17
Pharmacokinetics in Healthy Volunteers
The pharmacokinetics and pharmacodynamics of riociguat in humans were first evaluated in 118 healthy volunteers during two safety and tolerability studies. The first was a randomized, placebo-controlled, single-dose study that included 58 healthy white male patients. Riociguat doses during this study ranged from 0.25 to 5 mg.17
A multidose phase I study evaluated riociguat in 60 healthy male volunteers for 10 days. Patients were randomized to receive either riociguat 0.5 mg thrice daily (TID), 1 mg TID, 1.5 mg TID, 2.5 mg twice daily or TID, or placebo.
The results from both the studies are tabulated below:
|
Single dose study |
Multiple Dose Study |
|
Riociguat was extensively absorbed, with a time to maximum plasma concentration (Tmax) of 0.5–1.5 hours and demonstrated dose-proportionality and linear pharmacokinetics. The mean half-life of riociguat was 6.8 + 5 hours and the mean half-life of the active M1 metabolite was 13.9 hours. The effect of riociguat on heart rate (HR), systolic blood pressure (SBP), and diastolic blood pressure (DBP) was dose-dependent, and was evident starting at riociguat doses of 1 mg.
|
The geometric mean for riociguat’s AUC accumulation ratio was 1.10–1.57 and for the maximum plasma concentration ( Cmax) accumulation ratio was 1.05–1.46, an expected finding given the dosing interval and terminal half-life. Riociguat increased heart rate from baseline in a dose dependent fashion on study day 1 which was still present on study day 10 for most groups, albeit to a lesser extent. A dose-dependent decrease in SBP and DBP was also observed on study day 1, but was present on study day 10 to a greater extent. Results suggested that the heart rate-increasing effects of riociguat are better tolerated over time, whereas systemic BP-lowering effects may be more enduring |
Pharmacokinetics in PAH and CTEPH
Absorption:
The pharmacokinetics of riociguat has been assessed in patients with PAH (n=13) and CTEPH (n=6).
- Riociguat demonstrated dose-proportional pharmacokinetics in patients with PAH and CTEPH, similar to healthy volunteers; however, clinically significant differences were observed in C max and AUC.
- Absorption was rapid, with a Tmax of 0.25–1.5 hours in both patients and healthy volunteers
- The volume of distribution in the 1.0 and 2.5 mg groups was 0.35 + 0.03 L/ kg and 0.38 + 0.08 L/kg, respectively.
- Half-life of Riociguat is 10-12 hours in PAH patients. 19
|
Subjects |
AUC mcg.hr/L |
Cmax mcg/L |
|
Healthy |
348.5 |
74 |
|
PAH patients |
1411 |
119.4 |
Patients with PAH had an approximately 3-fold higher dose-normalized AUC and 1.5- to -2- fold higher dose-normalized Cmax.
Absorption:
The absolute bioavailability of riociguat is high (94%).
Intake with food reduced riociguat AUC slightly; Cmax was reduced by 35%.
Bioavailability (AUC and Cmax) for riociguat administered orally as a crushed tablet suspended in applesauce or in water is comparable with that of a whole tablet.
Distribution
Plasma protein-binding in humans is high at approximately 95%, with serum albumin and alpha-1 acidic glycoprotein being the main binding components. The volume of distribution is moderate, with volume of distribution at steady state being approximately 30 L.
Biotransformation
N-demethylation, catalysed by cytochrome CYP1A1, CYP3A4, CYP3A5 and CYP2J2, is the major biotransformation pathway of riociguat, leading to its major circulating active metabolite M-1 (pharmacological activity: one-tenth to one-third of riociguat), which is further metabolised to the pharmacologically inactive N-glucuronide. Formation of the M1 is mainly catalyzed by CYP1A1. At therapeutic concentrations riociguat or M1 does not inhibit any of the major CYPs.
CYP1A1 catalyses the formation of riociguat’s main metabolite in the liver and lungs and is known to be inducible by polycyclic aromatic hydrocarbons, which, for example, are present in cigarette smoke.
Excretion
Total riociguat (parent compound and metabolites) is excreted via both renal (33–45%) and biliary/faecal routes (48–59%). Approximately 4–19% of the administered dose was excreted as unchanged riociguat via the kidneys. Approximately 9–44% of the administered dose was found as unchanged riociguat in faeces.
Use in Special Populations
Paediatric
The safety and efficacy of riociguat in children and adolescents below 18 years of age have not been established. No clinical data are available. Non-clinical data show an adverse effect on growing bone. Until more is known about the implications of these findings, the use of riociguat in children and in growing adolescents should be avoided.
Geriatric
In elderly patients (age 65 years or older) there is a higher risk of hypotension and, therefore, particular care should be exercised during individual dose titration.
Renal Impairment
Data in patients with severe renal impairment (creatinine clearance [Crcl] <30 mL/min) are limited and there are no data for patients on dialysis. Therefore, use of riociguat is not recommended in these patients. Patients with moderate renal impairment (Crcl <50–30 mL/min) showed a higher exposure to this medicine. There is a higher risk of hypotension in patients with renal impairment; therefore, particular care should be exercised during individual dose titration.
Hepatic Impairment
Patients with severe hepatic impairment (Child-Pugh C) have not been studied and, therefore, use of riociguat is contraindicated in these patients. Patients with moderate hepatic impairment (Child-Pugh B) showed a higher exposure to this medicine. Particular care should be exercised during individual dose titration.
Landmark Trials
Chronic Thromboembolic Pulmonary Hypertension Soluble Guanylate Cyclase–Stimulator Trial 1 (CHEST-1)
Ghofrani HA, D'Armini AM, Grimminger F, et al Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. CHEST-1 Study Group. N Engl J Med. 2013, 369(4):319-29
Study objective: Evaluation of efficacy and safety of riociguat treatment in patients with inoperable CTEPH or persistent/ recurrent pulmonary hypertension after pulmonary endarterectomy.
Study design: Prospective, Phase 3, 16 week, randomized double-blind, placebo controlled, multicenter, international study.
Inclusion criteria:
- Patients (n=261) with inoperable CTEPH, or persistent/recurrent PH after surgery
- CTEPH diagnosed using > 2 imaging methods (V/Q scan, Pulmonary angiogram, MRI)
- 6MWD> 150m to < 450m, PVR>300 dyn.s-1. cm-5, mPAP> 25mm Hg
Exclusion criteria:
- Patients receiving an ERA, prostacyclin analogue, PDE-5 inhibitor, or NO donor < 3 months before study
During the initial 8-week dose-adjustment phase, riociguat was adjusted every 2 weeks according to an individual dose-adjustment scheme based on the patient’s systolic blood pressure and signs or symptoms of hypotension (Fig. 9).
.jpg?updated=20230105112149)
The starting dose was 1 mg three times daily. Riociguat dose was:
- Increased (by 0.5 mg three times daily) if trough systolic blood pressure was ≥95 mmHg
- Maintained if systolic blood pressure was 90 to 94 mmHg
- Decreased (by 0.5 mg three times daily) if systolic blood pressure was <90 mmHg without symptomatic hypotension.
Riociguat treatment was temporarily discontinued if systolic blood pressure was <90 mmHg with clinical symptoms of hypotension (e.g., presyncope or dizziness, as assessed by the individual investigator) and then restarted after 24 hours with a 0.5 mg reduced dose.
A decrease from the starting dose of 1 mg three times daily to 0.5 mg three times daily was permitted. Patients in the placebo group underwent sham adjustment, following the same adjustment regimen as the riociguat group.
The doses reached at the end of the 8-week adjustment phase were the appropriate dose for the patient, and the patient continued taking the drug at that dose for another 8 weeks. Patients were seen at weeks 2, 4, 6, and 8 (during the dose-adjustment phase) and then at weeks 12 and 16 (during the maintenance phase).10
Primary end point: was the change from baseline to the end of week 16 in the distance walked in 6 minutes.
Secondary end points: included changes from baseline in
- Pulmonary vascular resistance (PVR),
- N-terminal pro–brain natriuretic peptide (NT-proBNP) level,
- World Health Organization (WHO) functional class,
- Time to clinical worsening,
- Borg dyspnea score,
- Quality-of-life variables, and
- Safety
Results
PRIMARY END POINT
At week 16, the 6-minute walk distance had increased from baseline by a mean of 39 m in the riociguat group, as compared with a mean decrease of 6 m in the placebo group (least-squares mean difference, 46 m; 95% confidence interval [CI], 25 to 67; P<0.001). (Fig 10)
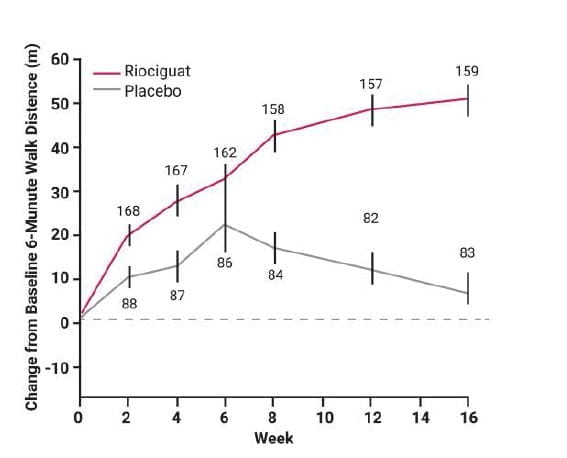
Mean (±SE) changes from baseline in the distance walked in 6 minutes during the 16-week study. The number at each data point indicates the number of patients included in the assessment at that time point. The least-squares mean difference in the distances at week 16 was 46 m (95% CI, 25 to 67; P<0.001). The last observed value (not including follow-up) was carried forward for patients who completed the study or withdrew; the worst value (0 m) was imputed in the case of death or clinical worsening without a termination visit or without a measurement at the termination visit.
SECONDARY END POINTS
PVR decreased by -226 dyn · sec · cm–5 in the riociguat group, as compared with an increase of +23 dyn · sec · cm–5 in the placebo group (least-squares mean difference, –246 dyn · sec · cm–5; 95% CI, –303 to –190; P<0.001)
Riociguat was also associated with significant improvement in other hemodynamic variables, including mean pulmonary-artery pressure (mPAP) and cardiac output (Table 2). Levels of NT-proBNP were significantly reduced in patients treated with riociguat, and changes in WHO functional class (fig 11) at 16 weeks also significantly favored riociguat.
|
Secondary end-points |
Placebo (change from baseline) Mean (+ SD) |
Riociguat (change from baseline) Mean (+ SD) |
P value |
|
Pulmonary vascular resistance (dyn.sec.cm-5) |
23 (+ 274) |
-226 (+ 248) |
<0.001 |
|
NT-proBNP (pg/ml) |
76 (+ 1447) |
-291 (+ 1717) |
<0.001 |
|
Borg dyspnea score |
0.2 (+ 2.4) |
-0.8 (+ 2) |
0.004 |
|
5Q-5D |
-0.08 (+ 0.34) |
0.06 (+ 0.28) |
<0.001 |
|
Hemodynamic variable |
|||
|
Mean pulmonary arterial pressure (mmHg) |
-0.3 (+ 11.8) |
-9 (+ 12) |
<0.001 |
|
Cardiac out put |
-0.03 (+ 1.07) |
0.8 (+ 1.1) |
<0.001 |
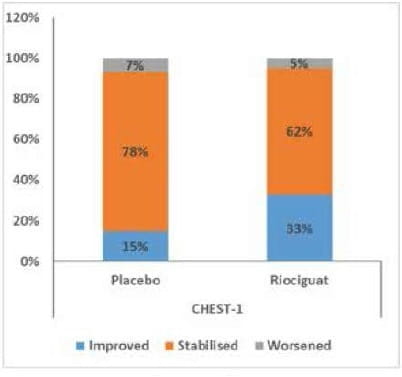
There was no significant difference in the incidence of clinical-worsening events between the riociguat and placebo groups (2% and 6%, respectively; P = 0.17) (Fig 12)
|
Conclusion: Riociguat significantly improved exercise capacity in patients with CTEPH who were deemed to be ineligible for surgery or who had persistent or recurrent pulmonary hypertension after undergoing pulmonary endarterectomy. Significant improvements were also observed in the clinically relevant secondary end points, including pulmonary vascular resistance, NT-proBNP level, and WHO functional class, cardiac output and mean arterial pressure. |
Riociguat for the Treatment of Chronic Thromboembolic Pulmonary Hypertension: A Long-term Extension Study (CHEST-2)
Simonneau G, D'Armini AM, Ghofrani HA, et al Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2).Eur Respir J. 2015, 45:1293-302
Study objective: Evaluation of long-term safety and tolerability of riociguat treatment in patients with CTEPH.
Study design: Open labelled 1-year extension, multi-center, prospective.
Inclusion criteria: Patients satisfied the CHEST-1 inclusion criteria and those who completed the 16-week double blind CHEST-1 trial. All the eligible patients received open-label riociguat individually adjusted up to a maximum dose of 2.5 mg three times daily irrespective of whether they had received riociguat or placebo in CHEST-1. 10
Exclusion criteria: Patients with riociguat-related SAEs from CHEST-1.
Primary endpoint: Safety and tolerability of riociguat.
Secondary efficacy endpoints:
- 6-min walking distance (6MWD)
- World Health Organization (WHO) functional class (FC).
Results
Of the 243 patients who completed CHEST-1, 237 (98%) entered CHEST-2, 73% patients had inoperable CTEPH and 27% had persistent/recurrent PH followed by PEA.
At the end of the 8-week double-blinded dose-adjustment phase, most patients in the former riociguat and placebo groups were receiving riociguat 2.5 mg three times daily (82% and 90%, respectively). This was sustained for up to 1 year, when 90% of patients were receiving riociguat 2.5 mg (maximum dose) three times daily.
During the open-label study phase of CHEST-2, 23 patients received at least one up-titration of riociguat dose and 13 patients received at least one down-titration.
At the start of CHEST-2, all patients were receiving riociguat monotherapy. Of patients treated for 1 year in CHEST-2, 145 (92%) out of 157 were continuing to receive monotherapy and 12 (8%) patients were receiving additional PH-specific medication [eight (5%) were receiving ERAs and four (3%) were receiving prostanoids].
Primary end-point
- Riociguat was well-tolerated; 5% of patients withdrew due to AEs.
- Hypotension was reported in 6% of patients and syncope in 7%.
- Drug related AEs were reported in 46% of patients, the most common were dizziness (10%), dyspepsia (8%) and hypotension (5%).
- The most common drug related SAEs were syncope (2%) and hypotension (1%), which were resolved in all cases.
- Other adverse events: The most common AEs in CHEST-2 were nasopharyngitis, dizziness, peripheral oedema, diarrhea, cough, dysnpneoa, upper respiratory tract infection, hypotension and syncope.
- By 1 year, majority of patients (90%) were receiving riociguat 2.5 mg t.i.d.
- Infrequent dose changes were observed during the open-label phase of the study, which supports the long-term tolerability of this treatment.
Secondary end point analysis
6MWD
Improvements in 6MWD in the riociguat group of CHEST-1 were sustained at week 12 and year 1 of CHEST-2 (Fig 13).
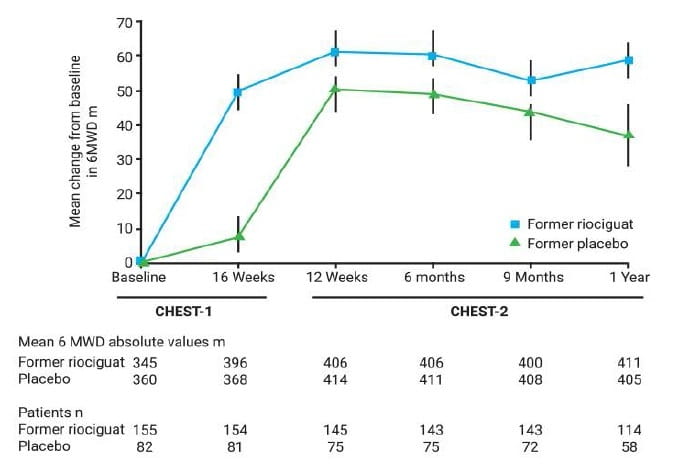
In the former riociguat group, mean±SD 6MWD had changed from CHEST-1 baseline by +50±59 m at week 16 of CHEST-1 (n=154) and by +61±59 m at week 12 of CHEST-2 (n=145; total 28 weeks).
In the former placebo group, a marginal change in 6MWD was observed at week 16 of CHEST-1 (+8±63 m; n=81); however, a change of +51±64 m (n=75) was observed at week 12 of CHEST-2 following transition to riociguat, which is comparable to the former riociguat group (Fig.11).
At 1 year of CHEST-2, 6MWD had changed by +59±58 m for the former riociguat group (n=114), +37 ±69 m in the former placebo group (n=58) and +51±62 m in the overall population (n=172) (fig. 11). In the overall population, mean ±SD 6MWD was 409±96 m (n=172) at 1 year, compared with 351±78 m (n=237) at baseline.
NT-proBNP
The decreases in NT-proBNP in the riociguat groups of CHEST-1 were sustained for up to 1 year in CHEST-2. Patients in the placebo group of CHEST-1 showed a similar decrease in NT-proBNP following transition to riociguat in CHEST-2. At 1 year in the overall population, NT-proBNP changed by −416±1321 pg·mL−1 (n=149) versus baseline (n=204).
WHO FC
At 1 year, WHO FC had improved/stabilised/worsened in 50/45/4%, respectively, of patients in the former riociguat group (n=117), 39/59/2% of patients in the former placebo group (n=59) and 47/50/3% of patients in the overall population (n=176) (Fig 14). The proportion of patients in the overall population in WHO FC I/II/III/IV at 1 year was 14/54/31/1% (n=177), respectively, compared with 1/31/65/3% (n=236) at baseline. (Fig 14)
.jpg?updated=20230105112731)
Survival and clinical worsening
o 16% of patients experienced a clinical worsening event during CHEST-2.
o The estimated rate of clinical worsening-free survival at 1 year was 88% (95% CI 83–92%) and (Fig 15).
o The estimated overall survival rate at 1 year of CHEST-2 was 97%.
.jpg?updated=20230105112837)
Quality of life
The improvement in Borg dyspnoea score seen in the former riociguat group at the end of CHEST-1 (n=154) was maintained in CHEST-2 at week 12 (n=145) and year 1 (n=113), while patients in the former placebo group showed minimal change at the end of CHEST-1 (n=81), with improved scores after the switch to riociguat in CHEST-2 at week 12 (n=75) and year 1 (n=58). Likewise, improvement in EQ-5D questionnaire score seen in the former riociguat group at the end of CHEST-1 (n=153) was maintained in CHEST-2 at week 12 (n=147) and year 1 (n=114), while patients in the former placebo group showed no improvement during CHEST-1 (n=80), but showed moderate improvement after the switch to riociguat in CHEST-2 at week 12 (n=76) and year 1 (n=59). A similar trend was observed for LPH, although this end-point was not measured beyond week 8 of CHEST-2.
|
|
Former Riociguat |
Former Placebo |
||
|
Quality of life |
Chest-1 week 16 (baseline) |
Chest-2, 1 year |
Chest-1 week 16 (baseline) |
Chest-2, 1 year |
|
Borg Dyspnea score |
-1.05 + 2.26 |
-0.8 + 2.41 |
-0.06 + 2.15 |
-0.57 + 1.98 |
|
EQ-5D* |
0.1 + 0.23 |
0.12 + 0.29 |
-0.03+ 0.26 |
0.01 + 0.30
|
|
* Euro Quality of life 5 Dimensions self assessment questionnaire score: range from -0.6-1.0, higher score indicates better quality of life. |
||||
|
IN SUMMARY
CONCLUSION Long-term treatment with riociguat showed a favourable benefit–risk profile in patients with inoperable CTEPH or persistent/recurrent PH after PEA and the study supports the use of riociguat as a long-term treatment for patients with inoperable or persistent/recurrent CTEPH. |
Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator Trial 1 (PATENT-1)
Ghofrani HA, Galiè N, Grimminger F, Grünig E, et al Riociguat for the treatment of pulmonary arterial hypertension.N Engl J Med. 2013;369(4):330-40.
Study objective: Evaluation of the efficacy and safety of riociguat treatment in patients with PAH
Study design: Multicenter, prospective, phase 3, randomised double-blind study, placebo controlled study trial
Total duration of study 12 weeks
Treatment: 1. Oral riociguat individually dose-adjusted up to 2.5mg t.i.d.
2. The 8-week dose adjustment phase followed by 4-week maintenance phase. Patients were started with 1mg t.i.d and then up-titrated by 0.5 mg t.i.d at the interval of 2 weeks to reach maximum dose of 2.5 mg t.i.d. Same steps were taken in placebo group with mock up-titrations.
Maintenance phase included next 4 weeks after.
Patients:
Inclusion criteria:
- Patients (N=443) symptomatic pulmonary arterial hypertension, aged 18-80 years, (Idiopathic 61%, familial 2%, associated PAH due to connective tissue diseases 25%, congenital heart disease 8%, portal hypertension with liver cirrhosis 3%, or anorexigen/amphetamine use 1%)
- Treatment naïve patients (50%) and patients pre-treated with ERA or prostacyclin analogue (except IV) (50%)
- 6MWD > 150m to < 450m
- PVR >300 dyn. s-1. com-5
- mPAP > 25mmHg
- WHO-FC I: 3%, WHO-FC II: 42%, WHOFC III: 54%, WHO-FC IV: 1%
Exclusion criteria:
- Patients receiving PDE-5 inhibitors
- Pulmonary hypertension seen in severe COPD, uncontrolled arterial hypertension, left heart failure
Patients who were receiving no other treatment for pulmonary arterial hypertension (50%) and patients who were receiving endothelin-receptor antagonists or (nonintravenous) prostanoids (50%) were eligible.
Primary end point: Change from baseline to the end of week 12 in the distance walked in 6 minutes.
Secondary end points included the change in
- pulmonary vascular resistance, cardiac output, mPAP
- N-terminal pro–brain natriuretic peptide (NT-proBNP) levels,
- World Health Organization (WHO) functional class,
- time to clinical worsening,
- score on the Borg dyspnea scale,
- quality-of-life variables, and safety.
Results
Primary end point
At week 12, the 6-minute walk distance had increased from baseline by a mean of 30 m in the 2.5 mg–maximum group and had decreased by a mean of 6 m in the placebo group (least-squares mean difference, 36 m; 95% confidence interval [CI], 20 to 52; P<0.001(Fig. 16).

Fig 16. Mean Change from Baseline in the 6-Minute Walk Distance. Mean (±SE) changes from baseline in the distance walked in 6 minutes during the 12-week PATENT-1 study period are shown in the group that received riociguat at a dose up to 2.5 mg three times daily as compared with the placebo group. The data were analyzed in the modified intention-to-treat population without imputation of missing values; imputed values are provided at week 12. The number at each data point indicates the number of patients included in the assessment at that time point. The least-squares mean difference in the 6-minute walk distance at week 12 was 36 m (95% CI, 20 to 52; P<0.001). The last observed value (not including follow-up) was carried forward for patients who completed the study or withdrew; the worst value (0 m) was imputed in the case of death or clinical worsening without a termination visit or without a measurement at the termination visit.
Secondary end points in the 2.5 mg–maximum Group Versus the Placebo Group
Significant improvements and benefits were seen in riociguat group, as compared with the placebo group, with respect to other hemodynamic variables and secondary end points, NT-proBNP levels, WHO functional class, score on the Borg dyspnea scale and Quality of life questionnaire -Living with Pulmonary Hypertension (LPH).
Pulmonary vascular resistance Cardiac Index
.jpg?updated=20230105113058)
Table 4: Secondary end points (change from the baseline) in riociguat group compared to placebo group.
|
Secondary end-points |
Placebo (change from baseline) |
Riociguat (change from baseline) |
P value |
|
PVR (dyn.sec.cm-5) |
-9 + 317 |
-223 + 260
|
<0.001 |
|
NT-proBNP (pg/ml) |
232 + 1011 |
-198 + 1721
|
<0.001 |
|
Borg dyspnea score |
0.1 + 2.1 |
-0.4+ 1.7
|
0.002 |
|
QoL: LPH score* |
0.4 + 18.2 |
-6 + 18 |
0.002 |
|
Hemodynamic variable |
|||
|
Mean pulmonary arterial pressure (mmHg) |
-1 + 13 |
-9 + 11 |
<0.001 |
|
Cardiac output |
-0.01 + 1.07 |
1+ 1
|
<0.001 |
|
* LPH: Living with Pulmonary hypertension score: range from 0-105, with higher scores indicating worse quality of life. |
|||

Sub group analysis:
- In the study population, 50% patients received no treatment and 50 % received treatment for PAH (endothelin-receptor antagonist and prostanoids). Riociguat administration improved the 6MWD in both the sub-groups.
- Majority of the patients in PATENT-1 originally belonged to WHO FC II (42%) and III (54%). Patients in functional class III or IV had a significantly greater benefit with riociguat therapy than did those in functional class I or II.
|
Conclusion: Riociguat significantly improved exercise capacity in patients with pulmonary arterial hypertension. This benefit was consistent in patients who were receiving endothelin-receptor antagonists or prostanoids and in those who were receiving no other treatment for the disease. Riociguat also significantly and consistently improved a range of secondary efficacy end points, including pulmonary hemodynamics, WHO functional class, and time to clinical worsening. |
Pulmonary Arterial Hypertension Soluble Guanylate Cyclase–Stimulator Trial – The Long-term Extension Study (PATENT-2)
Rubin LJ, Galiè N, Grimminger F, Grünig E, et al Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2).Eur Respir J. 2015 May;45(5):1303-13
Study objective: Evaluation of safety and tolerability of long-term riociguat treatment in patients with symptomatic PAH
Study design: Prospective, Phase 3 study, multicenter, international (conducted in 97 centres across 27 countries), open labeled, long term extension study.
Treatment: Oral riociguat individually dose adjusted upto 2.5mg t.i.d
Primary objective: To assess the safety and tolerability of riociguat
Exploratory efficacy assessments: 6MWD and World Health Organization (WHO) functional class.
Inclusion criteria:
Patients (N=396) who have completed the 12 week, double-blind PATENT-1 trial and who satisfied the PATENT-1 criteria.
Exclusion criteria: Patients with ongoing riociguat-related SAEs from PATENT-1.
Patients:
- Of 405 patients who completed PATENT-1, 396 (98%) entered PATENT-2 study.
- At the end of the 8 week double blinded dose adjustment period, the majority of patients in the former riociguat 2.5mg group, former riociguat 1.5mg max group and placebo group received riociguat at a dose of 2.5mg, three times a day (29%, 91%, 77% respectively) in PATENT-2.
- This dose was sustained for 1 year for majority of patients in PATENT-2 study.
- At the start of PATENT-2, 50% patients were on additional PAH specific medication;
o 43% on endothelin receptor antagonist
o 6% non-intravenous prostanoids,
o 1% on both ERA and prostanoids.
Results
Mean treatment duration was 95 weeks (median 91 weeks) and cumulative treatment exposure was 718 patient-years.
At one year of PATENT-2, 164 (54%) were on additional PAH specific medication; 140 (46%) on ERAs, 11 (4%) on prostanoids, 13 (4%) on both ERS and prostanoids.
Safety and tolerability:
o Riociguat was well tolerated, 10% patients withdrew due to AEs. The most common drug related AEs were dizziness, headache, and dyspepsia.
o Hypotension was reported in 9% of patients and syncope in 7%.
o Drug-related AEs were reported in 54% of patients; the most common were dizziness (9%), headache (8%) and dyspepsia (8%).
o SAEs were reported in 52% of patients, and drug-related SAEs in 7%.
o The most common drug-related SAEs included syncope and worsening PAH. All cases were resolved, except for two cases of worsening PAH (one fatal and one not resolved).
o Riociguat was well-tolerated both in treatment-naïve patients as well as in patients who were already being treated with an ERA or a prostanoid.
o The good tolerability of riociguat was underlined by the fact that the majority of patients were receiving the maximum 2.5 mg three times daily dose of riociguat at 1 year.
6MWD
Improvements in 6MWD in the riociguat groups of the PATENT-1 study were sustained at week 12 and at 1 year of the PATENT-2 study, and patients in the placebo group of the PATENT-1 study showed comparable improvements with the former riociguat groups following transition to riociguat (Fig 19).
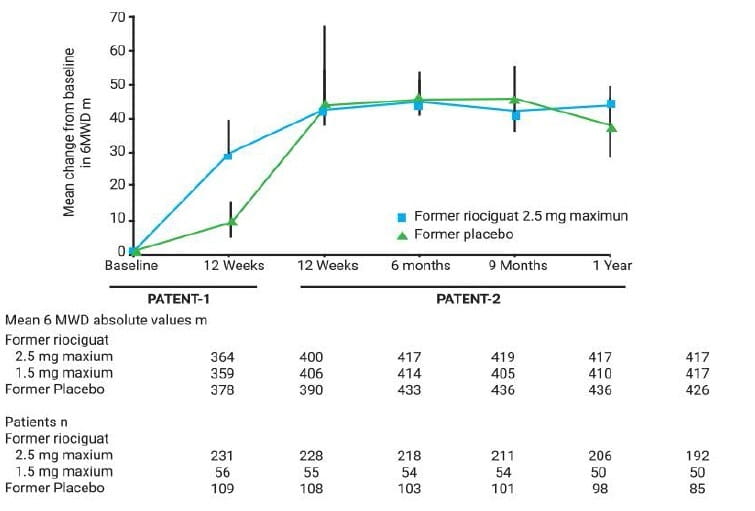
At 1 year of the PATENT-2 study, 6MWD had improved by mean±SD 53±70 m for the former riociguat 2.5 mg maximum group (n=192), 56±88 m in the former riociguat 1.5 mg maximum group (n=50), 46±76 m in the former placebo group (n=85) and 51±74 m in the overall population (n=327) (fig. 19).
In the overall population, the absolute mean±SD 6MWD was 419±97 m (n=327) at 1 year, compared with 367±67 m (n=396) at baseline.
Riociguat increased the proportion of patients achieving a 6MWD ⩾380 m in the PATENT-1 study, and this persisted for up to 1 year in the PATENT-2 study. The proportion of former placebo patients achieving a 6MWD ⩾380 m showed a similar increase following transition to riociguat in the PATENT-2 study.
NT-proBNP
The decreases in NT-proBNP in the riociguat groups of the PATENT-1 study were sustained for up to 1 year in the PATENT-2 study, with former placebo patients showing a similar decrease following transition to riociguat. In the overall population the decrease in NT-proBNP was -284+ 1653 pg.ml-1.
WHO functional class
At 1 year, WHO functional class had improved, stabilised or worsened in: 36%, 57% and 7% of patients, respectively, in the former riociguat 2.5 mg maximum group (n=199); 31%, 67% and 2% of patients in the former riociguat 1.5 mg maximum group (n=51); 26%, 66% and 8% of patients in the former placebo group (n=89); and 33%, 61% and 6% of patients in the overall population (n=339). The proportion of patients in WHO functional classes I, II, III and IV in the overall population was 8%, 61%, 28% and 2% (n=339) at 1 year, compared with 3%, 43%, 54% and 1% (n=395) at baseline.(Fig 20)
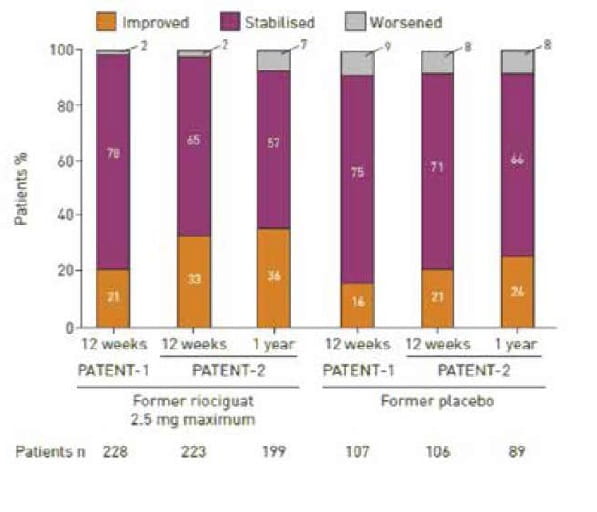
Fig 20 The proportion of patients in whom World Health Organization functional class was improved, stabilised or worsened in the former riociguat 2.5 mg maximum patient group, the former riociguat 1.5 mg maximum patient group and the former placebo patient group. Data are observed values. Percentages may not add up to 100% due to rounding.
Survival and clinical worsening
o 21% of patients experienced a clinical worsening event during the PATENT-2 study.
o Clinical worsening-free survival at 1 year was 88%.
o Estimated survival rate at 1 year was 97% . (Fig 21).
o Survival was also similar between treatment-naïve patients and patients pretreated with ERAs or prostanoids
.jpg?updated=20230105113519)
Quality of life: Improvements in EQ-5D and Borg dyspnoea scores in patients receiving riociguat in the PATENT-1 study were sustained in the overall PATENT-2 study population for up to 1 year (Table 5). Improvements in LPH score in the riociguat 2.5 mg maximum and the 1.5 mg maximum groups in the PATENT-1 study continued through to week 8 of the PATENT-2 study (LPH was not measured beyond week 8 during PATENT-2). However, a minimal change was observed in the former placebo group at week 8 during the PATENT-2 study.
|
At 1 year of PATENT-2 |
Former riociguat 2.5 mg t.i.d max dose |
Former riociguat 1.5 mg t.i.d max dose |
Former placebo |
|
Borg dyspnea score |
-0.52 + 1.98 (n=192) |
-0.18 + 1.58 (n=49) |
-0.45 + 2.06 (n=84) |
|
EQ-5D |
0.06 +0.24 (n=193) |
0.13 + 0.24 (n=50) |
0.07 + 0.20 (n=83) |
Sub-group analysis
- Patients in study population included idiopathic PAH (62%), followed by CTD-PAH (24%), congenital heart disease (8%), portopulmonary hypertension (3%), familial (2%) and drug- and toxin- induced (1%). Sub group analysis showed that survival data was similar in patients with idiopathic/familial PAH and PAH associated with connective tissue disease.
|
SUMMARY AND CONCLUSION
|
Survival was also similar between treatment-naïve patients and patients pretreated with ERAs or prostanoids, with estimated 1-year survival rates of 97% in both subgroups.
RESPITE: Switching to Riociguat in Pulmonary Arterial Hypertension Patients with Inadequate Response to Phosphodiesterase-5 Inhibitors
Hoeper MM, Simonneau G, Corris PA, et al. RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase-5 inhibitors. Eur Respir J 2017; 50: 1602425
A proportion of pulmonary arterial hypertension (PAH) patients do not reach treatment goals with phosphodiesterase-5 inhibitors (PDE5i). RESPITE investigated the safety, feasibility and benefit of switching from PDE5i to riociguat in these patients.
Study objective: To evaluate safety, feasibility and clinical benefits to replace PDE-5i therapy with riociguat in patients with PAH (who demonstrate insufficient response to PDE-5 inhibitors).
Study design: Prospective, Phase IIIb study, 24 week, single arm, open label, multicenter, international, study.
Treatment:
o Oral riociguat individually dose-adjusted up to 2.5 mg t.i.d
o 8 week dose adjustment phase followed by a 16 week maintenance phase
Study duration: RESPITE was a 24-week, open-label, multicentre, uncontrolled study.
Inclusion criteria:
- Patients (aged 18-75 yrs) (N=61) with idiopathic, familial, drug/toxin-induced or associated PAH due to CHD, demonstrating insufficient response to the treatment despite PDE-5i therapy for > 3 months.
- Patients with or without stable ERA therapy.
- Patients in WHO FC III,
- Patients with 6MWD 165–440 m,
- Patients with cardiac index <3.0 L·min−1·m−2 and pulmonary vascular resistance >400 dyn·s·cm−5 underwent a 1–3 day PDE5i treatment free period before receiving riociguat adjusted up to 2.5 mg maximum t.i.d.
Exclusion criteria:
- Previous treatment with riociguat
- Concomitant use of PAH-specific medications were not allowed during the study (PDE-5i: sildenafil, tadalafil, NO donors: nitrates)
- Treatment with prostacyclin analogues within last 90 days
- Other groups of PH and evidence of clinically significant restrictive or obstructive lung diseases
- DLCO<30% predicted
- History of serious hemoptysis
- Renal insufficiency
- Hepatic dysfunction,
Secondary end-points: included change in 6MWD, WHO FC, N-terminal prohormone of brain natriuretic peptide (NT-proBNP) and safety.
Of 61 patients enrolled, 51 (84%) completed RESPITE, 50 (82%) were receiving concomitant ERA.
Results
6MWD
In patients completing 24 weeks’ treatment, mean±SD 6MWD increased from baseline by +31±63 m (95% CI 13–49 m; p=0.0010; Fig 22).
.jpg?updated=20230105113621)
NT-proBNP
NT-proBNP levels decreased significantly by 347±1235 pg·mL−1 (Mean±SD), a relative reduction of 22% (95% CI 5−37%; p=0.0170; fig 23).
.jpg?updated=20230105113721)
WHO FC
At week 24, 28/52 (54%) patients had improved WHO FC (52% to WHO FC II, 2% to WHO FC I; p<0.0001; Fig 24).
.jpg?updated=20230105113820)
Fig 24 World Health Organization functional class (WHO FC) at baseline (the last documented value while still receiving phosphodiesterase-5 inhibitor), week 12 and week 24 in RESPITE. Data are observed values. *: p<0.0001, change from baseline to weeks 12 and 24; #: n=52 due to one patient withdrawal at day +159 having undergone a week 24 assessment of WHO FC.
Hemodynamic variables
At week 24, mean±SD PVR was reduced by -103±296 dyn·s·cm–5 (95% CI –188 to –18 dyn·s·cm–5; p=0.0184), mean±SD mPAP was reduced by -2.8±8.8 mmHg and right atrial pressure was reduced by –0.8±4.2 mmHg. Mean±SD cardiac index and mixed venous oxygen saturation increased by +0.3±0.5 L·min−1·m−2 (95% CI 0.2– 0.5 L·min−1·m−2; p=0.0001) and +1.0±6.3%, respectively.
|
Parameters (at 24 week) |
Change from baseline |
|
PVR |
-103 + 296 dyn·s·cm–5 |
|
mPAP |
-2.8 + 8.8 mmHg |
|
Right atrial pressure |
–0.8 + 4.2 mmHg |
|
cardiac index |
+0.3 + 0.5 L·min−1·m−2 |
|
Mixed venous oxygen saturation |
+1.0 + 6.3 % |
Adverse events
|
Summary RESPITE was the first study to investigate potential clinical improvement by switching therapies that target different molecules in the same signalling pathway in PAH and has provided preliminary evidence that this approach may be not only tolerable and safe, but also potentially beneficial in PAH patients with an insufficient response to their previous treatment, which consisted of PDE5i monotherapy or PDE5i/ERA combination therapy. Conclusion The findings from RESPITE indicate that replacing PDE5i with riociguat may be a feasible and effective treatment strategy for patients with PAH who have an insufficient clinical response to PDE5i monotherapy or to combination therapy with ERA and PDE5i. |
During the study, 58 patients (95%) experienced an adverse event. Four patients (7%) experienced adverse events leading to discontinuation of study drug, including two patients (3%) with right ventricular failure (days +15 and +158 after starting riociguat treatment), one patient (2%) with asthenia (day +2) and one patient (2%) with symptomatic hypotension (day +16). Of the two patients experiencing right ventricular failure, one concurrently experienced renal failure and asymptomatic hypotension, and the other concurrently experienced dyspnoea.
Riociguat as a Bridge Therapy to Balloon Pulmonary Angioplasty in Patients with Severe Chronic Thromboembolic Pulmonary Hypertension
Medical therapy for CTEPH has been also used as bridging treatment for those patients who await surgery (pulmonary endarectomy). However, there are very limited studies to conclude the effectiveness of bridging treatment. 25-27
Riociguat has been in one study used as a bridging treatment to balloon pulmonary angioplasty.28
The study included 27 consecutive patients with inoperable, severe CTEPH, who had undergone BPA earlier. Severe CTEPH patients were defined by WHO-FC III or IV and mean pulmonary arterial pressure (mPAP) of more than 30mmHg.
Nineteen patients received riociguat (6.5±1.5mg) before BPA (Riociguat Group), and the remaining eight patients did not any medications (No Pre-Med Group). The change of hemodynamic parameters and the plasma brain natriuretic peptide (BNP) levels, the numbers of BPA session, the complications associated with BPA, and the total amount of the contrast agent and radiation exposure in both groups were assessed.
End point of BPA was assumed to be that condition when mPAP<25mmHg or further BPA was judged to be difficult to perform.
Results:
Plasma BNP and mPAP levels significantly improved in the Riociguat Groups before BPA 1st session but not in No Pre-Med Group. There were no adverse effects associated with Riociguat.
Table 6a: Effect of Riociguat on Plasma BNP concentration and mPAP levels
|
1st session |
Riociguat Group |
No Pre-Med Group |
||
|
|
Base line |
After BPA |
Base line |
After BPA |
|
plasma BNP levels |
188.5±204.8 pg/ml
|
71.4±69.2 pg/ml |
146.9±164.1 pg/ml , |
141.9±147.8 pg/ml |
|
p value |
p<0.01 |
|
p=0.8 |
|
|
mPAP |
from 43.1±4.2 mm Hg
|
34.5±5.5 mmHg |
from 41.1±4.0 to mmHg |
43.1±9.8 mmHg, |
|
p value |
p<0.001 |
|
p=0.5 |
|
At the endpoint of BPA, the plasma BNP levels and mPAP levels was improved in both groups.
Table 6b: Plasma BNP concentration and mPAP levels at end point.
|
|
Riociguat Group |
No Pre-Med Group |
|
plasma BNP levels |
from 188.5±204.8 to 33.9±32.7 pg/ml, |
from 146.9±164.1 to 24.1±7.4 pg/ml |
|
|
p<0.01 |
p=0.07 |
|
mPAP |
43.1±4.2 to 21.5±2.0 mmHg |
from 41.1±4.0 to 22.9±2.4 mmHg |
|
|
p<0.001 |
p<0.001 |
The plasma BNP levels and mPAP at the baseline and endpoint between were no significant differences in both groups. The rate of severe complications associated with BPA was not significantly different in both groups (5.9% in Riociguat Group vs 11.1% in No Pre-Med Group).
Importantly, the total numbers of BPA session and the total amount of contrast agent by which the mPAP was achieved to be less than 25 mmHg were significantly less in Riociguat Group than No Pre-Med Group (Riociguat Group vs No Pre-Med Group: total numbers of BPA session; 2.7±1.2 vs 4.5±1.8, p<0.01, total amount of contrast agent; 570±263 ml vs 871±348 ml, p<0.05).
These findings indicate that Riociguat pre-medical treatment could serve as a bridge therapy to effectively perform BPA procedure in patients with inoperable severe CTEPH.
Safety and Tolerability of Riociguat
Riociguat was well tolerated in the clinical studies.
The short- and long-term safety of riociguat was assessed during the PATENT and CHEST trials, which included 789 and 429 patient-years of follow up, respectively. Treatment-emergent adverse events reported during the PATENT and CHEST trials are shown below:17
Table 7: Pooled Treatment-Emergent Adverse events from Phase 3 studies of Riociguat
|
Adverse events (AE) |
Placebo |
Riociguat |
|
Treatment emergent AE of special interest |
|
|
|
Hypotension |
2.8% |
9.6% |
|
Syncope |
3.7% |
1.6% |
|
Presyncope |
0.5% |
1.6% |
|
Hemorrhagic event |
0% |
2.4% |
|
Common Treatment emergent adverse events (TEAE ) |
|
|
|
Headache |
17.3% |
26.2% |
|
Dyspepsia |
7.9 % |
18.5 % |
|
Peripheral edema |
15.0 % |
16.6 % |
|
Nausea |
10.7 % |
13.8 % |
|
Dizziness |
12.1 % |
18.5 % |
|
Diarrheoa |
7.9 % |
12.2 % |
|
Nasopharingitis |
10.3 % |
12.2 % |
|
Vomiting |
6.5 % |
10.1% |
- A riociguat plasma concentration-response relationship was observed for hypotension in patients with baseline SBP ≤ 110 mm Hg.
- Higher prevalence of hypotension was observed in the following cases receiving riociguat :
o For patients ≥ 75 years old (29.5%) -compared to placebo (10%),
o For patients with renal impairment (Creatinine Clearance [CrCL] 30-49ml/min). There is no data in patients with CrCL <30ml/min (31.6%)- compared to placebo (8.3%)
- 74% of hypotensive events in the riociguat 2.5 mg maximum group occurred after 2 days.
- There is concern regarding serious adverse events with riociguat.
- Serious renal adverse events occurred in four patients receiving riociguat (~0.8%) and none in those receiving placebo.
- There is a concern regarding potential nephrotoxicity of the M1 metabolite based on preclinical data; however, there was no clear relationship between these rare events, riociguat, or hypotension associated with treatment.
Indications, Dosage and Administration
Indications
- Chronic-Thromboembolic Pulmonary Hypertension
Riociguat is indicated for the treatment of adults with persistent/recurrent chronic thromboembolic pulmonary hypertension (CTEPH), (WHO Group 4) after surgical treatment, or inoperable CTEPH, to improve exercise capacity and WHO functional class.
- Pulmonary Arterial Hypertension
Riociguat is indicated for the treatment of adults with pulmonary arterial hypertension (PAH), (WHO Group 1), to improve exercise capacity, WHO functional class and to delay clinical worsening.
Dosage and Administration
Recommended Dosage in Adult Patients
For non-hypotensive patients:
- Recommended starting dosage is 1 mg taken 3 times a day
- Up-titrate the dose by 0.5 mg taken three times a day
- Highest tolerated dosage, up to a maximum of 2.5 mg taken three times a day.
For hypotensive patients:
- Recommended starting dose of 0.5 mg taken three times a day
- If systolic blood pressure remains greater than 95 mmHg and the patient has no signs or symptoms of hypotension, dose increase should be no sooner than 2 weeks apart. If at any time, the patient has symptoms of hypotension, decrease the dosage by 0.5 mg taken three times a day.
Warnings and Precautions
- Pregnancy (Boxed warning)
Riociguat carries a boxed warning indicating that the drug can harm a developing fetus and should not be used in pregnant women. Women taking riociguat must comply with pregnancy testing requirements and be counseled regarding the importance of contraception use while taking the medication.
Pregnancy testing in females of reproductive potential
Initiate treatment with RIOTEPH Tablets in females of reproductive potential only after a negative pregnancy test. Obtain a monthly pregnancy test during treatment.
- Hypotension: Because riociguat reduces blood pressure, hypotension and ischemia may occur in patients with hypovolemia, severe left ventricular outflow obstruction, resting hypotension, autonomic dysfunction and those who take concomitant antihypertensive or strong cytochrome P and P-glycoprotein/breast cancer resistance protein inhibitors.
- Bleeding: During clinical trials of riociguat, serious bleeding occurred in 2.4% of patients in the riociguat group and in none of the patients in the placebo group. Serious hemoptysis was observed in five (1%) patients taking riociguat and in none of the patients receiving placebo, with one event resulting in death. Other serious hemorrhagic events included two patients with vaginal hemorrhage, two patients with catheter-site haemorrhage and one patient each with subdural hematoma, hematemesis and intra-abdominal haemorrhage.
- Pulmonary Veno-Occlusive Disease
Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD). Therefore, administration of riociguat to such patients is not recommended. Should signs of pulmonary edema occur, the possibility of associated PVOD should be considered and, if confirmed, discontinue treatment with riociguat.
- Contraindications:
- The concomitant use of riociguat is contraindicated with any PDE inhibitors, including the specific PDE-5 inhibitors sildenafil, tadalafil and vardenafil, and nonspecific PDE inhibitors, including dipyridamole and theophylline. The potentially unfavorable safety signals with sildenafil plus riociguat (increased prevalence of hypotension), and no evidence of a positive benefit to risk ratio, mean that concomitant administration of riociguat with PDE5 inhibitors is contraindicated
- Riociguat concomitant use is also contraindicated with nitrates in any form (including NO donors, such as amyl nitrite).
- Riociguat is contraindicated is contraindicated in patients with pulmonary hypertension associated with idiopathic interstitial pneumonias (PH-IIP). In patients with PH-IIP, riociguat was associated with increased SAEs and mortality, and an unfavourable risk: benefit ratio.
Place of Riociguat in Therapy
- Riociguat represents a significant advancement in the medical management of CTEPH as the first FDA-approved medication for CTEPH.
- For patients who are ineligible for or do not want to undergo PEA, have inoperable disease, or have recurrent or persistent CTEPH after PEA, riociguat is a safe and effective option.
- For PAH non-respondents to PDE-5i and ERA, riociguat can serve as an effective alternative therapy.
Summary
- Chronic thromboembolic pulmonary hypertension (CTEPH) is an insidious disease with a poor prognosis.
- Pulmonary thromboendarterectomy (PEA) is the first line treatment of choice for CTEPH, with a real potential for cure.
- Patients should be evaluated at specialized centers for PEA eligibility.
- Riociguat, a soluble guanylate cyclase stimulator, is the first medical therapy approved by the US FDA for patients with inoperable CTEPH or persistent pulmonary hypertension after PEA.
- In CHEST 1 - a large, multicenter, placebo-controlled trial, riociguat up to 2.5 mg three-times a day, significantly improved exercise capacity, hemodynamics, functional class, quality of life and markers of right ventricular load.
- In the long-term open-label study -CHEST 2, which followed the randomized trial, riociguat was associated with maintenance of improvements in exercise capacity, hemodynamics, functional class and survival.
- Side effects associated with riociguat include hypotension and bleeding (especially in patients on chronic anticoagulation).
Prescribing Information
Riociguat
WARNING: EMBRYO-FETAL TOXICITY
|
Composition
Rioteph 0.5mg Tablets
Each film coated tablet contains:
Riociguat……………………………………0.5mg
Colour: Titanium Dioxide IP
Rioteph 1 mg Tablets
Each film coated tablet contains:
Riociguat……………………………………1.0mg
Colour: Titanium Dioxide IP & Ferric Oxide USP-NF Yellow
Rioteph 1.5 mg Tablets
Each film coated tablet contains:
Riociguat……………………………………1.5mg
Colour: Titanium Dioxide IP & Ferric Oxide USP-NF Yellow
Dosage Form
Tablets
Pharmacology
Pharmacodynamics
Mechanism of Action
Riociguat is a stimulator of soluble guanylate cyclase (sGC), an enzyme in the cardiopulmonary system, and the receptor for nitric oxide (NO). When NO binds to sGC, the enzyme catalyses synthesis of the signalling molecule, cyclic guanosine monophosphate (cGMP). Intra-cellular cGMP plays an important role in regulating processes that influence vascular tone, proliferation, fibrosis and inflammation.
Pulmonary hypertension is associated with endothelial dysfunction, impaired synthesis of NO, and insufficient stimulation of the NO-sGC-cGMP pathway.
Riociguat has a dual mode of action. It sensitises sGC to endogenous NO by stabilising the NO-sGC binding. Riociguat also directly stimulates sGC independently of NO.
Riociguat restores the NO-sGC-cGMP pathway and leads to increased generation of cGMP.
Pharmacodynamic effects
Riociguat restores the NO-sGC-cGMP pathway, resulting in a significant improvement of pulmonary vascular haemodynamics and an increase in exercise ability.
There is a direct relationship between riociguat plasma concentration and haemodynamic parameters such as systemic and pulmonary vascular resistance, systolic blood pressure and cardiac output.
Pharmacokinetics
Absorption
The absolute bioavailability of riociguat is high (94%). Riociguat is rapidly absorbed, with maximum concentrations (Cmax) appearing 1–1.5 hours after tablet intake. Intake with food reduced riociguat AUC slightly; Cmax was reduced by 35%.
Bioavailability (AUC and Cmax) for riociguat administered orally as a crushed tablet suspended in applesauce or in water is comparable with that of a whole tablet.
Distribution
Plasma protein-binding in humans is high at approximately 95%, with serum albumin and alpha-1 acidic glycoprotein being the main binding components. The volume of distribution is moderate, with volume of distribution at steady state being approximately 30 L.
Biotransformation
N-demethylation, catalysed by cytochrome CYP1A1, CYP3A4, CYP3A5 and CYP2J2, is the major biotransformation pathway of riociguat, leading to its major circulating active metabolite M-1 (pharmacological activity: one-tenth to one-third of riociguat), which is further metabolised to the pharmacologically inactive N-glucuronide.
CYP1A1 catalyses the formation of riociguat’s main metabolite in the liver and lungs and is known to be inducible by polycyclic aromatic hydrocarbons, which, for example, are present in cigarette smoke.
Excretion
Total riociguat (parent compound and metabolites) is excreted via both renal (33–45%) and biliary/faecal routes (48–59%). Approximately 4–19% of the administered dose was excreted as unchanged riociguat via the kidneys. Approximately 9–44% of the administered dose was found as unchanged riociguat in faeces.
Based on in vitro data, riociguat and its main metabolite are substrates of the transporter proteins, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). With a systemic clearance of about 3–6 L/h, riociguat can be classified as a low-clearance drug. Elimination half-life is about 7 hours in healthy subjects and about 12 hours in patients.
Linearity
Riociguat pharmacokinetics are linear from 0.5 to 2.5 mg. Inter-individual variability (CV) of riociguat exposure (AUC) across all doses is approximately 60%.
Use in Special Populations
Paediatric
The safety and efficacy of riociguat in children and adolescents below 18 years of age have not been established. No clinical data are available. Non-clinical data show an adverse effect on growing bone. Until more is known about the implications of these findings, the use of riociguat in children and in growing adolescents should be avoided.
Geriatric
In elderly patients (age 65 years or older) there is a higher risk of hypotension and, therefore, particular care should be exercised during individual dose titration.
Renal Impairment
Data in patients with severe renal impairment (creatinine clearance [Crcl] <30 mL/min) are limited and there are no data for patients on dialysis. Therefore, use of riociguat is not recommended in these patients. Patients with moderate renal impairment (Crcl <50–30 mL/min) showed a higher exposure to this medicine. There is a higher risk of hypotension in patients with renal impairment; therefore, particular care should be exercised during individual dose titration.
Hepatic Impairment
Patients with severe hepatic impairment (Child-Pugh C) have not been studied and, therefore, use of riociguat is contraindicated in these patients. Patients with moderate hepatic impairment (Child-Pugh B) showed a higher exposure to this medicine. Particular care should be exercised during individual dose titration.
Indications
- Chronic Thromboembolic Pulmonary Hypertension
Rioteph tablets is indicated for the treatment of adults with persistent/recurrent chronic thromboembolic pulmonary hypertension [CTEPH] (WHO Group 4) after surgical treatment, or inoperable CTEPH, to improve exercise capacity and WHO functional class.
In CTEPH, pulmonary endarterectomy is the treatment of choice as it is a potentially curative option. According to standard medical practice, expert assessment of operability should be done prior to treatment with Rioteph tablets.
- Pulmonary Arterial Hypertension (PAH)
Rioteph tablets is indicated for the treatment of adults with PAH (WHO Group 1), to improve exercise capacity, WHO functional class, and to delay clinical worsening.
In PAH, studies with Rioteph tablets have been mainly performed in forms related to idiopathic or heritable PAH and PAH associated with connective tissue disease. The use of Rioteph tablets in other forms of PAH not studied is not recommended.
Dosage and Administration
Recommended Dosage in Adult Patients
The recommended starting dosage is 1 mg taken three times a day. For patients who may not tolerate the hypotensive effect of Rioteph tablets, consider a starting dose of 0.5 mg taken three times a day. If systolic blood pressure remains greater than 95 mmHg and the patient has no signs or symptoms of hypotension, up-titrate the dose by 0.5 mg taken three times a day. Dose increases should be no sooner than 2 weeks apart. The dose can be increased to the highest tolerated dosage, up to a maximum of 2.5 mg taken three times a day. If at any time, the patient has symptoms of hypotension, decrease the dosage by 0.5 mg taken three times a day.
Dosage Interruption
If a dose is missed, advise patients to continue with the next regularly scheduled dose. In case Rioteph is interrupted for 3 days or more, re-titrate Rioteph tablets.
Pregnancy Testing in Females of Reproductive Potential
Obtain pregnancy tests prior to initiation and monthly during treatment.
Use in Patients Who Smoke
Consider titrating to dosages higher than 2.5 mg three times a day, if tolerated, in patients who smoke. A dose decrease may be required in patients who stop smoking.
Strong CYP and P-gp/BCRP Inhibitors
Consider a starting dose of 0.5 mg, three times a day, when initiating Rioteph tablets in patients receiving strong CYP450 and P-gp/BCRP inhibitors such as azole antimycotics (e.g. ketoconazole, itraconazole) or HIV protease inhibitors (e.g. ritonavir). Monitor for signs and symptoms of hypotension on initiation and on treatment with strong CYP and P-gp/BCRP inhibitors.
Transitioning to and from Riociguat
- Discontinue sildenafil at least 24 hours prior to administering Rioteph tablets.
- Discontinue tadalafil at least 48 hours prior to administering Rioteph tablets. Consider initiating Rioteph tablets at a starting dose of 0.5 mg in patients at risk of hypotension. It is recommended to monitor for signs and symptoms of hypotension on initiation.
- Discontinue Rioteph tablets at least 24 hours prior to administering a phosphodiesterase type 5 (PDE5) inhibitor. It is recommended to monitor for signs and symptoms of hypotension on initiation.
Contraindications
Pregnancy
Riociguat may cause foetal harm when administered to a pregnant woman. Riociguat is contraindicated in females who are pregnant. Riociguat was consistently shown to have teratogenic effects when administered to animals. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the foetus.
Phosphodiesterase Inhibitors (PDE5 inhibitors)
Concomitant administration of riociguat with specific PDE5 inhibitors (such as sildenafil, tadalafil or vardenafil) or nonspecific PDE5 inhibitors (such as dipyridamole or theophylline) is contraindicated. Do not administer within 24 hours of sildenafil. Do not administer 24 hours before or within 48 hours after tadalafil.
Pulmonary Hypertension Associated with Idiopathic Interstitial Pneumonias (PH-IIP)
Riociguat is contraindicated in patients with pulmonary hypertension associated with idiopathic interstitial pneumonias (PH-IIP).
Severe Hepatic Impairment (Child-Pugh C)
Riociguat is contraindicated in patient with severe hepatic impairment (Child-Pugh C).
Hypersensitivity
Riociguat is contraindicated in patients with hypersensitivity to the active substance or to any of the excipients.
Nitrates or Nitric Oxide Donors
Co-administration of riociguat with nitrates or nitric oxide donors (such as amyl nitrite) in any form, including recreational drugs called ‘poppers’, is contraindicated.
Systolic Blood Pressure
Riociguat is contraindicated in patient with systolic blood pressure <95 mmHg at treatment initiation.
Warnings and Precautions
Drug Interactions
Pharmacodynamic Interactions
Nitrates: Co-administration of riociguat with nitrates or NO donors (e.g. amyl nitrite) in any form is contraindicated because of hypotension.
PDE Inhibitors: Co-administration of riociguat with specific PDE5 inhibitors (e.g. sildenafil, tadalafil, or vardenafil) and nonspecific PDE inhibitors (e.g. dipyridamole or theophylline), is contraindicated because of hypotension. Do not administer within 24 hours of sildenafil. Do not administer 24 hours before or within 48 hours after tadalafil. Clinical experience with co-administration of riociguat, and other PDE inhibitors (e.g. milrinone, cilostazole, roflumilast) is limited.
Pharmacokinetic Interactions
Smoking: Plasma concentrations in smokers are reduced by 50–60% compared with nonsmokers. Based on pharmacokinetic modelling, doses higher than 2.5 mg three times a day may be considered for patients who are smokers in order to match the exposure seen in nonsmoking patients. Safety and effectiveness of riociguat doses higher than 2.5 mg three times a day have not been established. A dose reduction should be considered in patients who stop smoking.
Strong CYP and P-gp/BCRP Inhibitors: Concomitant use of riociguat with strong CYP inhibitors and Pgp/BCRP inhibitors such as azole antimycotics (e.g. ketoconazole, itraconazole) or HIV protease inhibitors (e.g. ritonavir) increase riociguat exposure and may result in hypotension. Consider a starting dose of 0.5 mg three times a day when initiating riociguat in patients receiving strong CYP and P-gp/BCRP inhibitors. Monitor for signs and symptoms of hypotension on initiation and on treatment with strong CYP and P-gp/BCRP inhibitors. A dose reduction should be considered in patients who may not tolerate the hypotensive effect of riociguat.
Strong CYP3A Inducers: Strong inducers of CYP3A (e.g. rifampin, phenytoin, carbamazepine, phenobarbital or St. John’s wort) may significantly reduce riociguat exposure. Data are not available to guide dosing of riociguat when strong CYP3A inducers are co-administered.
Antacids: Antacids such as aluminium hydroxide/magnesium hydroxide decrease riociguat absorption and should not be taken within 1 hour of taking riociguat.
Renal Impairment
Data in patients with severe renal impairment (CRcl <30 mL/min) are limited and there are no data for patients on dialysis; therefore, riociguat is not recommended in these patients. Patients with mild and moderate renal impairment were included in the pivotal studies. There is increased riociguat exposure in these patients. There is a higher risk of hypotension in these patients; hence, particular care should be exercised during individual dose titration.
Hepatic Impairment
There is no experience in patients with severe hepatic impairment (Child-Pugh C); riociguat is contraindicated in these patients. PK data show that higher riociguat exposure was observed in patients with moderate hepatic impairment (Child-Pugh B). Particular care should be exercised during individual dose titration.
There is no clinical experience with riociguat in patients with elevated liver aminotransferases (>3 x upper limit of normal [ULN]) or with elevated direct bilirubin (>2 x ULN) prior to initiation of treatment; hence, riociguat is not recommended in these patients.
Pregnancy
Riociguat may cause foetal harm when administered to a pregnant woman and is contraindicated during pregnancy. Therefore, female patients at potential risk of pregnancy must use an effective method of contraception. Monthly pregnancy tests are recommended.
Pregnancy Category X
Riociguat was teratogenic and embryotoxic in rats at doses with exposures to unbound drug that were approximately 8 times and 2 times, respectively, the human exposure. In rabbits, riociguat led to abortions at 4 times the human exposure and foetal toxicity with exposures approximately 13 times the human exposure. If riociguat is used in pregnancy, or if the patient becomes pregnant while taking this drug, apprise the patient of the potential hazard to the foetus.
Lactation
It is not known if riociguat is present in human milk. Riociguat or its metabolites were present in the milk of rats. Because many drugs are present in human milk and because of the potential for serious adverse reactions in nursing infants from riociguat, discontinue nursing or riociguat.
Females and Males of Reproductive Potential
Female patients of reproductive potential must have a negative pregnancy test prior to starting treatment with riociguat, monthly during treatment, and 1 month after discontinuation of treatment with riociguat. Advise patients to contact their healthcare provider if they become pregnant or suspect pregnancy. Counsel patients on the risk to the foetus.
Contraception: Female patients of reproductive potential must use acceptable methods of contraception during treatment with riociguat and for 1 month after treatment with riociguat. Patients may choose one highly effective form of contraception (intrauterine devices [IUD], contraceptive implants or tubal sterilization) or a combination of methods (hormone method with a barrier method or two barrier methods). If a partner’s vasectomy is the chosen method of contraception, a hormone or barrier method must be used along with this method. Counsel patients on pregnancy planning and prevention, including emergency contraception, or designate counselling by another healthcare provider trained in contraceptive counselling.
Paediatric Use
The safety and efficacy of riociguat in children and adolescents below 18 years of age have not been established. No clinical data are available. Non-clinical data show an adverse effect on growing bone. Until more is known about the implications of these findings, the use of riociguat in children and in growing adolescents should be avoided.
Geriatric Use
In elderly patients (age 65 years or older), there is a higher risk of hypotension and, therefore, particular care should be exercised during individual dose titration.
Pulmonary Veno-Occlusive Disease
Pulmonary vasodilators may significantly worsen the cardiovascular status of patients with pulmonary veno-occlusive disease (PVOD). Therefore, administration of riociguat to such patients is not recommended. Should signs of pulmonary oedema occur, the possibility of associated PVOD should be considered and treatment with riociguat should be discontinued.
Respiratory Tract Bleeding
In PH patients, there is increased likelihood for respiratory tract bleeding, particularly among patients receiving anticoagulation therapy. A careful monitoring of patients taking anticoagulants according to common medical practice is recommended.
The risk of serious and fatal respiratory tract bleeding may be further increased under treatment with riociguat, especially in the presence of risk factors, such as recent episodes of serious haemoptysis, including those managed by bronchial arterial embolisation. Riociguat should be avoided in patients with a history of serious haemoptysis or who have previously undergone bronchial arterial embolisation. In case of respiratory tract bleeding, the prescriber should regularly assess the benefit-risk of treatment continuation.
Hypotension
Riociguat has vasodilatory properties, which may result in lowering of blood pressure. Before prescribing riociguat, physicians should carefully consider whether patients with certain underlying conditions, could be adversely affected by vasodilatory effects (e.g. patients on antihypertensive therapy or with resting hypotension, hypovolaemia, severe left ventricular outflow obstruction or autonomic dysfunction).
Riociguat must not be used in patients with a systolic blood pressure below 95 mmHg. Patients older than 65 years are at increased risk of hypotension. Therefore, caution should be exercised when administering riociguat in these patients.
Smokers
Plasma concentrations of riociguat in smokers are reduced compared with non-smokers. Dose adjustment may be necessary in patients who start or stop smoking during treatment with riociguat.
Concomitant Use with Other Medicinal Products
- The concomitant use of riociguat with strong multi-pathway CYP and P-gp/BCRP inhibitors such as azole antimycotics (e.g. ketoconazole, itraconazole) or HIV protease inhibitors (e.g. ritonavir) is not recommended, due to the pronounced increase in riociguat exposure.
- The concomitant use of riociguat with strong CYP1A1 inhibitors, such as the tyrosine kinase inhibitor, erlotinib, and strong P-gp/BCRP inhibitors, such as the immunosuppressive agent cyclosporine A, may increase riociguat exposure. These medicinal products should be used with caution. Blood pressure should be monitored and dose reduction of riociguat be considered.
Undesirable Effects
Summary of the Safety Profile
The safety of riociguat has been evaluated in Phase III studies of 681 patients with CTEPH and PAH receiving at least one dose of riociguat. Most of the adverse reactions are caused by relaxation of smooth muscle cells in the vasculature or the gastrointestinal tract.
The most commonly reported adverse reactions, occurring in ≥10% of patients under riociguat treatment (up to 2.5 mg three times daily), were headache, dizziness, dyspepsia, peripheral oedema, nausea, diarrhoea and vomiting. Serious haemoptysis and pulmonary haemorrhage, including cases with fatal outcome, have been observed in patients with CTEPH or PAH treated with riociguat.
The safety profile of riociguat in patients with CTEPH and PAH appeared to be similar and, therefore, adverse reactions identified from placebo-controlled 12- and 16-week clinical studies are presented as pooled frequency in the table below.
Adverse Reactions
The adverse reactions reported with riociguat are listed below by MedDRA system organ class and by frequency. Frequencies are defined as follows: very common (≥1/10), common (≥1/100 to <1/10) and uncommon (≥1/1,000 to <1/100).
Adverse Reactions Reported with Riociguat in the Phase III Studies
|
MedDRA System Organ Class |
Very Common |
Common |
Uncommon |
|
Infections and infestations |
|
Gastroenteritis |
|
|
Blood and the lymphatic system disorders
|
|
Anaemia (including respective laboratory parameters) |
|
|
Nervous system disorders
|
Dizziness Headache |
|
|
|
Cardiac disorders
|
|
Palpitations |
|
|
Vascular disorders
|
|
Hypotension |
|
|
Respiratory, thoracic and mediastinal disorders |
|
Haemoptysis Epistaxis Nasal congestion |
Pulmonary haemorrhage* |
|
Gastrointestinal disorders
|
Dyspepsia Diarrhoea Nausea Vomiting
|
Gastritis Gastro-oesophageal reflux disease Dysphagia Gastrointestinal and abdominal pains Constipation Abdominal distension |
|
|
General disorders and administration site conditions |
Oedema peripheral |
|
|
* Fatal pulmonary haemorrhage was reported in uncontrolled long-term extension studies
If case of any side effects, talk to your doctor or pharmacist or write to drugsafety@cipla.com. You can also report side effects directly via the National Pharmacovigilance Programme of India by calling on 1800 180 3024.
By reporting side effects, you can help provide more information on the safety of this product.
Overdosage
Inadvertent overdosing with total daily doses of 9–25 mg riociguat between 2 and 32 days was reported. Adverse reactions were similar to those seen at lower doses.
In case of overdose, standard supportive measures should be adopted as required. In case of pronounced hypotension, active cardiovascular support may be required. Based on the high plasma protein-binding, riociguat is not expected to be dialysable.
Incompatibility
Not applicable.
Storage and Handling Instructions
Do not store above 30°C.
Packaging Information
Rioteph 0.5 mg Tablets ........... Pack of 10 tablets
Rioteph 1 mg Tablets .............. Pack of 10 tablets
Rioteph 1.5 mg Tablets ........... Pack of 10 tablets
June 2018
References:
1. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A,et al 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015 Oct;46(4):903-75.
2. Madani M, Ogo T, Simonneau G. The changing landscape of chronic thromboembolic pulmonary hypertension management. Eur Respir Rev 2017; 26: 170105
3. Gall H, Hoeper MM, Richter MJ, et al. An epidemiological analysis of the burden of chronic thromboembolic pulmonary hypertension in the USA, Europe and Japan. Eur Respir Rev 2017; 26: 160121
4. Hoeper MM. Chronic thromboembolic pulmonary hypertension after acute pulmonary embolism: to screen or not to screen? Eur Respir J 2018; 51: 1800440
5. Simonneau G, Torbicki A, Dorfmüller P, et al. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur Respir Rev 2017; 26: 160112
6. Pengo V, Lensing AW, Prins MH, et al. Incidence of chronic thromboembolic pulmonary hypertension after pulmonary embolism. N Engl J Med 2004; 350: 2257–2264.
7. Pepke-Zaba J, Delcroix M, Lang I, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation 2011; 124: 1973–1981.
8. Lang IM. Chronic thromboembolic pulmonary hypertension – not so rare after all. N Engl J Med 2004; 350: 2236–2238.
9. Hoeper MM, Mayer E, Simonneau G, Rubin LJ. Chronic thromboembolic pulmonary hypertension. Circulation. 2006 Apr 25;113(16):2011-20.
10. Condliffe R, Kiely DG, Gibbs JS, et al. Prognostic and aetiological factors in chronic thromboembolic pulmonary hypertension. Eur Respir J 2009; 33: 332–338
11. Bonderman D, Wilkens H, Wakounig S, et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir J 2009; 33: 325–331.
12. Dutt, TS, Mohan, BVM et al Incidence of Chronic Thrombo-embolic Pulmonary Hypertension Following Acute Pulmonary Thrombo-embolism: An Indian Perspective Indian J Chest Dis Allied Sci 2013;55:205-207.
13. Kim NH, Lang IM. Risk factors for chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2012 Mar 1;21(123):27-31.
14. Lang IM, Dorfmüller P, Vonk Noordegraaf A The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann Am Thorac Soc. 2016 3: S215-21.
15. D'Armini AM Diagnostic advances and opportunities in chronic thromboembolic pulmonary hypertension. Eur Respir Rev. 2015 Jun;24(136):253-62.
16. Ghofrani HA, Humbert M, Langleben D, Riociguat: Mode of Action and Clinical Development in Pulmonary Hypertension. Chest. 2017 Feb;151(2):468-48
17. Makowski CT, Rissmiller RW, Bullington WM.Riociguat: a novel new drug for treatment of pulmonary hypertension.Pharmacotherapy. 2015 May;35(5):502-19.
18. DeSouza SA, Preston IR. The safety and effectiveness of riociguat to treat chronic thromboembolic pulmonary hypertension. Expert Rev Cardiovasc Ther. 2015 May;13(5):467-76.
19. Grimminger F, Weimann G, Frey R, Voswinckel R, Thamm M, Bölkow D First acute haemodynamic study of soluble guanylate cyclase stimulator riociguat in pulmonary hypertension. Eur Respir J. 2009 Apr;33(4):785-92.
20. Ghofrani HA, D'Armini AM, Grimminger F, et al Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. CHEST-1 Study Group. N Engl J Med. 2013, 369(4):319-29
21. Simonneau G, D'Armini AM, Ghofrani HA, et al Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: a long-term extension study (CHEST-2).Eur Respir J. 2015, 45:1293-302.
22. Ghofrani HA, Galiè N, Grimminger F, Grünig E, et al Riociguat for the treatment of pulmonary arterial hypertension.N Engl J Med. 2013;369(4):330-40.
23. Rubin LJ, Galiè N, Grimminger F, Grünig E, et al Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2).Eur Respir J. 2015 May;45(5):1303-13.
24. Hoeper MM, Simonneau G, Corris PA, et al. RESPITE: switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase-5 inhibitors. Eur Respir J 2017; 50: 1602425.
25. Reesink HJ, Surrie S et al Bosentan as a bridge to pulmonary endarterectomy for chronic thromboembolic pulmonary hypertensionJ Thorac Cardiovasc Surg 2010;139:85-91.
26. Nagaya N, Sasaki N et al Prostacyclin Therapy Before Pulmonary Thromboendarterectomy in Patients With Chronic Thromboembolic Pulmonary HypertensionCHEST 2003; 123:338–343.
27. Bresser P, Fedullo P et al Continuous intravenous epoprostenol for chronic thromboembolic pulmonary hypertension. Eur Respir J 2004; 23: 595–600.
28. Ueda, J et al Downloaded from https://academic.oup.com/eurheartj/article-abstract/38/suppl_1/ehx502.P2597/4089321, Accessed on 5 July, 2018