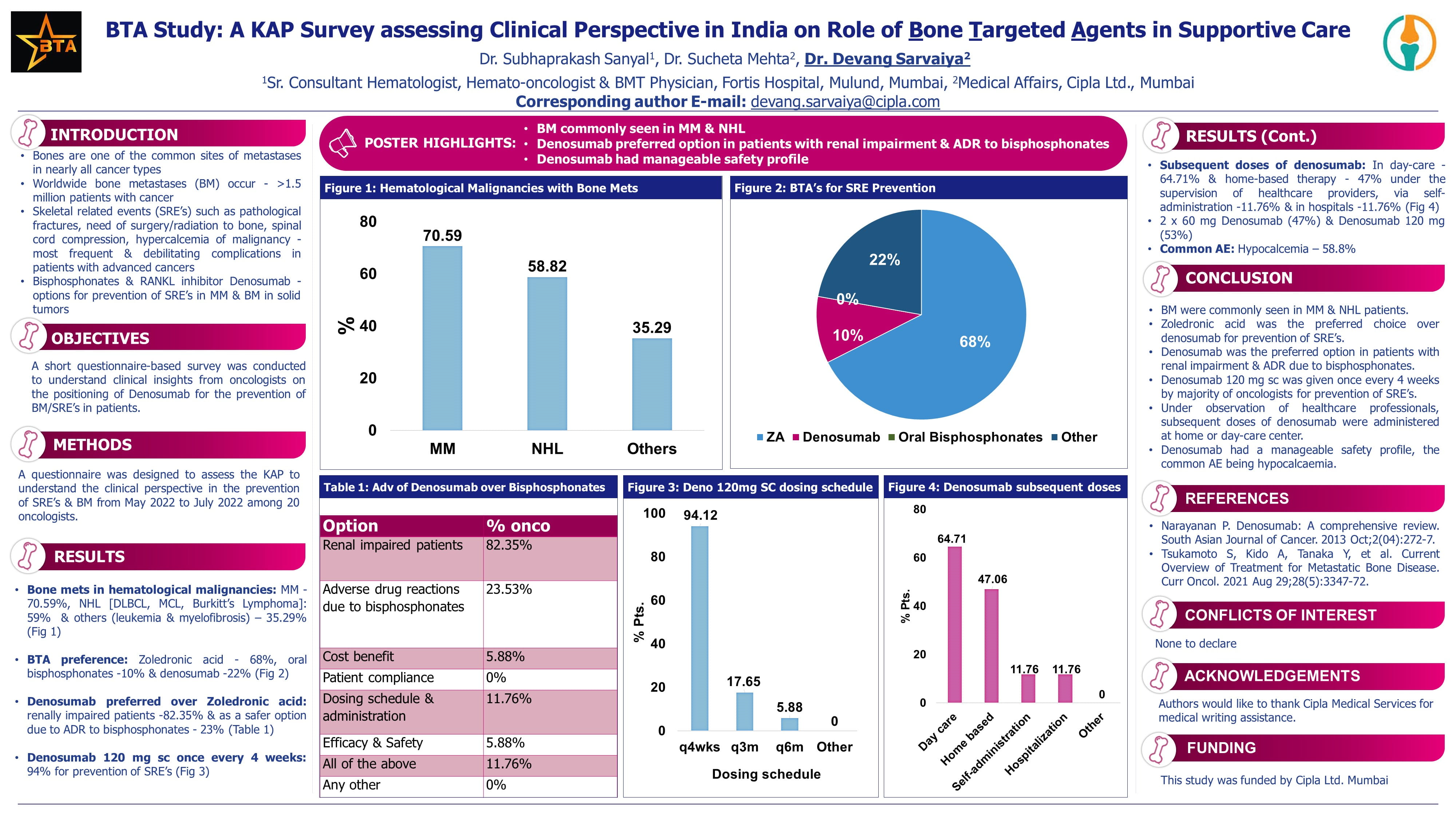
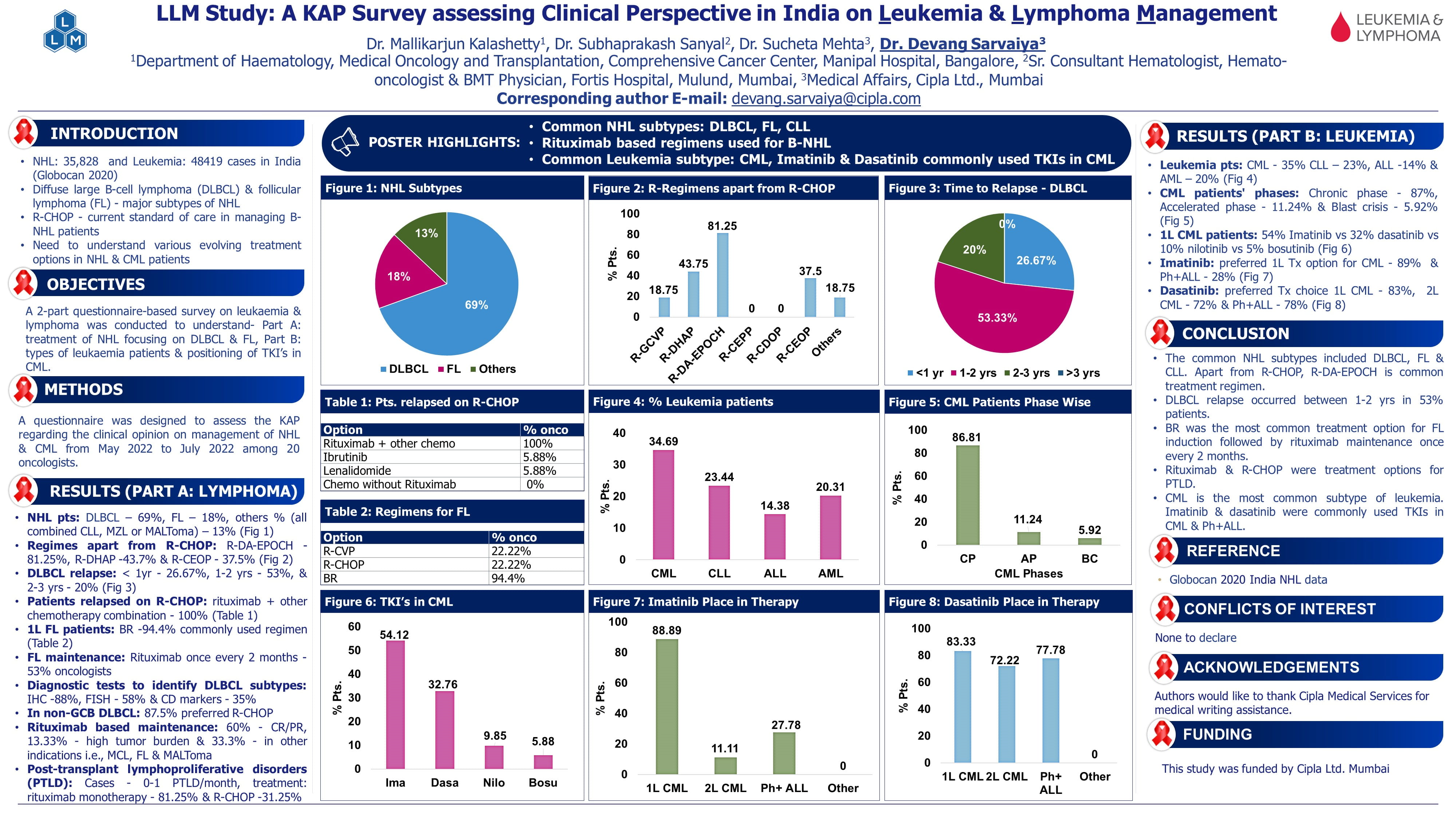
EHA 2023: Scientific Sessions Highlights






Targeted Therapy of Uncontrolled Erythrocytosis in Polycythemia vera with the Hepcidin mimetic, Rusfertide: - Blinded Randomized Withdrawal Results of the REVIVE
Study
Marina Kremyanskaya, MD, PhD, of the Icahn School of Medicine at Mount Sinai
Background
Polycythemia vera (PV) is a clonal myeloproliferative neoplasm characterized by uncontrolled erythrocytosis, systemic symptoms and an increased risk of thromboembolic (TE) and cardiovascular (CV) complications. Therapeutic phlebotomy (TP), with or without cytoreductive agents (CYTO) is used to control hematocrit (HCT) levels <45% to improve symptoms and decrease the risk of TE and CV complications. The previous study reported results demonstrating that adding rusfertide to prior PV treatments more optimally controls HCT levels and decreases the need for TP (EHA 2022). The present study report results from the randomized withdrawal phase (Part 2) which was unblinded and represents the primary objective of the REVIVE study.
Aims
To investigate the effect of rusfertide, a first-in-class hepcidin mimetic, on the control of erythrocytosis in patients with PV.
Methods
The REVIVE (PTG-300-04) trial (NCT04057040) consists of three stages. Eligibility criteria: diagnosis of PV (WHO 2016 criteria); ≥3 TPs in the 28 wk prior to enrollment with or without concurrent CYTO. Subcutaneous rusfertide was added to prior PV therapy. During Part 1 (28 wk), rusfertide dose was adjusted individually to control HCT <45%. During Part 2 (wk 29-41), the blinded randomized withdrawal phase, patients were randomized to either continue rusfertide or to matching placebo. A patient was defined as a responder if 3 criteria were met 1) HCT control without phlebotomy eligibility, 2) no TP, and 3) completed 12 wk of treatment. Need for TP was triggered by a) HCT ≥45% and ≥3% higher than wk 29 pre-randomization HCT or b) HCT>48% or c) ≥5% increase in HCT compared to wk 29 HCT. Responders and non-responders were allowed to participate in Part 3, a 3- yr open-label extension.
Results
-
53 subjects were randomized (27 placebo, 26 rusfertide) and completed part 2.
-
Demographics: Median age 58 yr (range, 27-77); 37 men (71.7%) 15 women (28.3%); concurrent therapy: PHL alone 29 (54.7%) PHL + CYTO 24 (45.3%).
-
Rusfertide met its primary efficacy end point of response as defined by not needing phlebotomy within 12 weeks (69.2% vs 18.5% in the placebo arm; P = .0003). Subgroup analysis showed superior efficacy for rusfertide relative to placebo in both concurrent therapy groups: TP alone (p=0.02) and TP+CYTO (p=0.02).
-
Rusfertide significantly improved maintenance of response, absence of the need for TP and persistent HCT control compared to placebo (p<0.0001).
-
In Part 1 the phlebotomy-free rate was 76.9% (wk 1-17) and 87.3% (wk 17-29).
-
In Part 2, the rate was 92.3% for rusfertide cohort.
-
Rusfertide was generally well tolerated; 83% of treatment-emergent adverse events (TEAEs) were grade 1-2, 17% were grade 3 with none grade 4 or 5.
-
Most common TEAEs were injection site reactions (ISRs), which were localized, and grade 1-2 in severity. ISRs decreased in incidence with continued treatment. Only 2 TEAEs led to treatment discontinuation.
-
Of the 70 patients enrolled, 52 (74.3%) have been treated for ≥1 year, 32 (45.7%) for ≥1.5 years, and 10 (14.3%) for ≥2 years, indicating long-term tolerability of rusfertide.
-
Rusfertide demonstrated favorable effects on several patient-reported outcomes, such as fatigue, problems with concentration, pruritus, and inactivity, and that was particularly seen in patients who had more severe symptoms at baseline.
Summary
The randomized withdrawal phase of the REVIVE study met its primary endpoint and demonstrated that rusfertide is a highly effective agent in patients with PV receiving TP with or without CYTO. Rusfertide is a novel hepcidin mimetic that selectively targets uncontrolled erythrocytosis in PV. Rusfertide is well tolerated and produces sustained and durable HCT control, obviating the need for TP in PV patients. Phase 3 VERIFY trial is ongoing to investigate rusfertide vs placebo, and there is a follow-on 2-year extension study opening this year for the patients who completed the REVIVE study
BMT-CTN 1506 (MORPHO): A Randomized Trial of the FLT3 Inhibitor Gilteritinib as Post-Transplant Maintenance for FLT3-ITD AML
Mark J. Levis, MD, PhD, of Johns Hopkins University School of Medicine
Background
Patients with acute myeloid leukemia with an internal tandem duplication mutation of FLT3 (FLT3-ITD AML) have a high risk of relapse and routinely undergo allogeneic hematopoietic cell transplantation (HCT). FLT3 inhibitors are often administered as post-HCT maintenance therapy to decrease relapse risk, but this practice is based on randomized studies of sorafenib that included patients salvaged with FLT3 inhibitors pre-transplant.
Aims
BMT-CTN1506 (“MORPHO”) was an international phase 3 randomized placebo-controlled, double blinded study of post-HCT maintenance with the FLT3 inhibitor gilteritinib. The primary objective was to determine if post-HCT maintenance with gilteritinib improved relapse-free survival (RFS) compared with placebo for participants (pts) with FLT3-ITD AML transplanted in first remission. The secondary objective was overall survival (OS). Additional secondary objectives included examining the effect of measurable residual disease (MRD) pre- and post-transplant on RFS and OS, rates of non-relapse mortality, event-free survival, and acute and chronic graft-versus-host disease (GVHD) in participants treated with gilteritinib versus placebo.
Methods
Adults with FLT3-ITD AML in first remission after receiving no more than two cycles of induction therapy with HCT planned within 12 months of achieving remission were screened for eligibility. After induction and any consolidation therapy, pts were registered and underwent HCT. After engraftment, between 30-90 days after HCT, they were randomized to placebo or 120 mg/day gilteritinib for 24 months. Marrow aspirates for MRD were collected pre-transplant, pre-randomization, and at 3, 6, 12, 18, and 24 months post-randomization. MRD was analyzed using a PCR-NGS assay that could detect a FLT3-ITD mutation at a level of 1 x 10-6. Randomization was stratified by pre-HCT MRD of 10-4 or greater, conditioning regimen intensity, and time from HCT to randomization of -/+ 60 days.
Results
-
The study included: screened 620, registered 488, and randomized 356 pts.
-
By intention-to-treat analysis, RFS was higher for pts randomized to gilteritinib, but the difference was not statistically significant (HR: 0.679; 95% CI: 0.459, 1.005; 2-sided p-value: 0.0518).
-
OS was similar in both groups (HR: 0.846; 95% CI: 0.554, 1.293; 2-sided p-value: 0.4394.
-
Two-year RFS was 77.2% (95% CI 70.1%, 82.8%) for gilteritinib and 69.9% (95% CI: 62.4%, 76.2%) for placebo. 50.6% of pts had MRD (10-6 or greater) pre-HCT or pre-randomization.
-
In pre-specified subgroup analysis, the effect of gilteritinib was more pronounced in pts with detectable MRD (HR=0.515, 95% CI: 0.316, 0.838, p = 0.0065) than in pts without detectable MRD (HR=1.213, 95% CI: 0.616, 2.387, p = 0.575). 143 (80.3%) in gilteritinib arm and 129 (72.9%) in placebo arm experienced dose interruptions and 97 (54.5%) in gilteritinib arm and 45 (25.4%) in placebo arm required dose reductions.
-
No new safety signals were observed, with the most common being nausea and differentiation syndrome, and there was encouraging clinical activity, with about one-third of participants achieving complete remission and a 45% overall response rate.
-
Treatment-emergent adverse events (TEAE), including neutrophil decrease (42.1 versus 15.8%) and chronic GVHD (52.2 versus 42.1%), were more common in the gilteritinib arm, as were TEAEs leading to withdrawal of treatment.
Summary
Gilteritinib appears to have a clear benefit for the 50% of pts with detectable MRD pre- or post-HCT, compared to those without detectable MRD. TEAEs associated with gilteritinib were primarily myelosuppression and increased incidence of chronic GVHD. The study results suggested that the data is among the first to support the effectiveness of MRD-based post-HCT maintenance therapy.
Cevidoplenib, A Selective Inhibitor of Spleen Tyrosine Kinase (SYK), in Persistent and Chronic Immune Thrombocytopenia (ITP): Efficacy and Safety in a Multicenter, Placebo controlled Phase 2 study
Jun Ho Jang, MD, PhD, of Samsung Medical Center in Seoul, Republic of Korea
Background
Immune thrombocytopenia (ITP), an autoimmune disorder is caused by auto-antibodies that target platelets, leading to a low platelet count. This can result in bruises and potentially life-threatening internal bleeding. There is an urgent need for a safer and more efficacious treatment option as many ITP patients are refractory to the current standard therapies including thrombopoietin-receptor agonists (TPO-RAs). Cevidoplenib (SKI-O-703) is a highly potent and selective inhibitor of spleen tyrosine kinase (SYK). By inhibiting the signaling downstream of B cell and Fc receptors, SYK inhibitor is expected to be effective in autoantibody-driven pathologies including ITP. Targeting SYK may be an effective way to increase platelet counts by reducing B cell-driven auto-antibody production as well as ameliorating macrophage-mediated platelet destruction. This phase 2 trial aimed to evaluate its performance on an end point of increasing platelet count past 30,000 per μL and doubling platelet count from baseline.
Aims
To evaluate the efficacy and safety of cevidoplenib in persistent and chronic ITP patients who are refractory to conventional therapy.
Methods
The efficacy and safety of oral cevidoplenib (CVP) were evaluated in the Phase II multicenter, randomized, double blind, placebo (PBO)-controlled, parallel dose trial (NCT04056195) in adults with persistent and chronic ITP who had failed to respond to or relapsed after prior therapy. Stable doses of background medications including corticosteroids and immunosuppressive drugs were permitted but the doses were fixed for at least 2 weeks before Day 1 and remained unchanged throughout the treatment period. Sixty-one participants were randomly assigned to 3 groups using a 2:2:1 ratio to receive 400 mg, 200mg CVP, and PBO twice daily for 12 weeks. The primary endpoint was the proportion of participants with platelet response, defined as platelet count ≥30,000/μL and doubling the baseline (average of 2 previous counts) without the use of rescue medication. Key secondary endpoints included the proportion of participants achieving pre-specified PLT count.
Results
-
Sixty-one participants were randomized (23 in 400mg CVP, 26 in 200 mg CVP, and 12 in PBO, respectively).
-
Participants had severe ITP (median baseline PLT of 8,500/μL) and 68.3% of the participants had received ≥3 previous lines of therapies.
-
Cevidoplenib was associated with a numerically greater rate of platelet response vs placebo (400 mg: 63.6%; 200 mg: 46.2%; placebo: 33.3%), but the difference was not statistically significant for either dose. Patients who sustained their platelet count, defined as having counts above 50,000 per μL at 4 or more of their last 6 visits, made up 27.3% of the 400 mg arm and 19.2% of the 200 mg arm; no patients in the placebo arm achieved this outcome.
-
Participants achieving 2 or more consecutive PLT counts ≥50,000/μL without the use of rescue medication were 40.9% on 400 mg CVP, 19.2% on 200 mg CVP vs. 8.3% on PBO (P=0.055 and P=0.371 for 400 mg and 200 mg CVP, respectively, vs. PBO).
-
Sustained PLT count (defined retrospectively as PLT counts ≥50,000/μL at ≥4 of the last 6 visits) was reached in 27.3% on 400 mg CVP and 19.2% on 200 mg CVP vs. 0% on PBO.
-
Mean change from baseline in the average of the last 2 available PLT counts (SD) were 41,600/μL (46,586) on 400 mg CVP, 36,020/μL (50,270) on 200mg CVP and 17,500/μL (26,821) on PBO.
-
Platelet responses to cevidoplenib were consistent regardless of prior TRO-RAs use or baseline PLT counts <15,000/μL.
-
Adverse events (AEs) were reported in 66.7% in the combined CVP groups (32 out of 48 participants treated with cevidoplenib) and 66.7% in the PBO group (8 out of 12 participants). The most frequently reported treatment related adverse events were ALT increase (8.3%), AST increase (6.3%) and nausea (4.2%). Most AEs were grade 1 or 2. Serious AEs were reported in 4.2% of the combined CVP group and 25% in the PBO.
-
Treatment related grade 3 or 4 AEs were observed in 6.3% participants in the combined CVP group and resolved spontaneously or with medical management. No death was observed.
Summary
Cevidoplenib 400 mg twice daily was generally well tolerated and demonstrated robust platelet responses in a significant proportion of participants who had failed multiple prior therapies. However further clinical studies in a larger number of participants for an extended period are required to confirm durability of the clinical benefits.
Activity, Tolerability, and Resistance Profile of the Menin Inhibitor Ziftomenib in Adults with Relapsed/Refractory NPM1-Mutated AML
Amir Fathi, MD, Massachusetts General Hospital, Boston
Background
The menin and histone-lysine-N-methyltransferase 2A (KMT2A) protein complex is an essential epigenetic regulator of genes critical for leukemogenesis in multiple leukemia subtypes, including in NPM1 mutant (NPM1m) acute myeloid leukemia (AML) and AML with KMT2A gene rearrangements (KMT2Ar). The presence of comutations can portend a poor prognosis, particularly in the relapsed/refractory (R/R) setting, which is an area of high unmet need.
Aims
The purpose of the Phase (Ph) 1 portion of KOMET-001 (NCT04067336) is to establish the safety and recommended phase 2 dose (RP2D) for ziftomenib monotherapy in NPM1m and KMT2Ar R/R AML.
Methods
KOMET-001 (NCT04067336) is a global, open-label Ph 1/2 study of ziftomenib in adult patients (pts) with R/R AML. The Ph 1 dose escalation and randomized dose expansion portion in pts with KMT2Ar or NPM1m R/R AML is fully enrolled. Ziftomenib is dosed orally, once daily, in 28-day cycles until relapse, progression, or unacceptable toxicity.
Results
-
Median age of RP2D pts was 70.5 years (22 to 86y).
-
FLT3 (30%) and IDH1/2 (40%) co-mutations were common (20% had both co-mutations).
-
Median number of prior therapies was 3.0 (r: 1 to 10); 20% had ≥1 prior stem cell transplant (SCT). Based on updated results, the complete remission (CR) rate for NPM1m pts at 600mg is 35%, with 40% of pts overall achieving composite CR (CRc) and ORR of 45%.
-
The median time to first response is 51 days (r: 26 to 225). One CR at the 200mg dose has an ongoing DoR of 35 cycles. The median DoR for all NPM1m pts achieving CRc is 8.2 months (m) per Kaplan-Meier estimate (95% CI: 1.0 to NE).
-
Two pts (1 CR and 1 CRi) underwent SCT and remain in remission as of the cutoff, one on post-SCT ziftomenib maintenance therapy. Molecular analyses suggest ziftomenib leads to measurable residual disease (MRD) clearance of target mutations such as NPM1, and co-mutations (eg, FLT3 and IDH1) likely by targeting the founding clone and subclonal events or through targeting aberrant gene expression, as at least 2 pts with FLT3 and IDH1 co-mutations present at baseline were undetectable after 2 cycles.
-
Most pts (85%) had at least one ≥Gr 3 treatment-emergent adverse event (TEAE); 30% were potentially treatment related. The most frequent (>20%) TEAEs ≥ Gr 3 were anemia (25%) and thrombocytopenia (20%).
-
Any grade differentiation syndrome (DS) was reported in 20%; most (n=3) were Gr 2. The resistance profile was explored and the resistance mutation MEN1-M327I was found to develop in 1 of 29 pts (3.4%), detected at C4D28; the pt maintained stable disease through cycle 7.
-
One key new biochemical finding, confirmed by crystal structure, demonstrates that ziftomenib retains full activity against the T349M mutation, detected in two thirds of pts who acquired menin gatekeeper mutations on another recent menin inhibitor trial.
Summary
Ziftomenib continues to demonstrate significant clinical activity in heavily pretreated and co-mutated R/R NPM1m AML pts where 35% of pts achieved CR. The safety profile remains consistent, and episodes of DS are clinically manageable. The study data reveals that remissions are durable, with MRD clearance of NPM1 and key co-mutations. Resistance mutations develop infrequently, and ziftomenib remains effective against a common menin gatekeeper mutation. A single-arm registration-directed Ph 2 study is currently accruing to further evaluate ziftomenib monotherapy in R/R NPM1m AML.
OMS906, A Mannan-binding Lectin-associated Serine Protease-3 (MASP-3) INHIBITOR, Normalizes Hemoglobin Levels in Treatment-naïve PNH patients: Interim Data from a Proof-of-concept Clinical Trial
Jens Panse, MD, University of Aachen, Germany
Background
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare and life-threatening blood disorder characterized by intravascular hemolysis (IVH), thrombophilia, and cytopenia. Terminal complement inhibition blocks IVH but inevitably leads to extravascular hemolysis (EVH). Proximal and alternative pathway (AP) inhibition block both IVH and EVH as demonstrated with C3, Factor B, and Factor D inhibitors. MASP-3 is a key activator of the AP, upstream of Factor D. OMS906 is a humanized mAb that binds to and inhibits MASP-3. In a Phase 1 study in healthy subjects, OMS906 was well tolerated with a long duration of MASP-3 inhibition.
Aims
To evaluate the safety and efficacy of OMS906 for PNH treatment in a proof-of-concept design.
Methods
This is an ongoing single-arm, open-label, clinical trial evaluating safety, PK, PD, and preliminary efficacy of OMS906 in adults with PNH. Patients are eligible for inclusion if they have a confirmed PNH diagnosis by flow cytometry; are complement inhibitor treatment-naïve or had an inadequate response to C5 inhibitors; and have baseline hemoglobin (Hgb) <10.5 g/dL. Patients receive 5 mg/kg subcutaneous (SC) OMS906 every 4 weeks, the lowest exposure cohort in this study. Primary endpoints are safety and tolerability. Secondary endpoints include efficacy (change from baseline Hgb and lactate dehydrogenase [LDH]); proportion of patients achieving Hgb ≥12 g/dL or increase in Hgb ≥2 g/dL; and proportion of transfusion-independent patients from Week 4 through Weeks 24 and 48. Planned enrollment for this cohort is approximately 10 patients.
Results
-
Nine treatment-naïve patients (5 male, mean age 43 years [range 27–72], 7 requiring pre-study transfusions) have been treated with OMS906. Coexisting conditions include aplastic anemia (n=2), iron deficiency (n=5), myelodysplastic syndrome (n=2), and chronic renal failure (n=3).
-
At baseline (N=9), mean Hgb and LDH were 6.78 g/dL and 1931 U/L. 8 patients have received 2 doses, 4 patients 3 doses, and 3 patients 5 doses. Four weeks after the first dose (n=8), mean Hgb increased by 3.43 g/dL (p=0.002) from a baseline of 6.33/6.20 g/dL (mean/median) and LDH fell by 1637 U/L (80%) (p<0.001) from a baseline of 2047/1573 U/L (mean/median).
-
At the latest timepoint of 16 weeks (n=3), mean Hgb increase was 9.70 g/dL (p=0.017) from a baseline of 6.13/6.40 g/dL (mean/median) and LDH reduction was 87% (p=0.003) from a baseline of 2251/2375 U/L (mean/median).
-
All patients (N=9) had an increase in Hgb >2 g/dL and all patients reaching 16 weeks have Hgb ≥15.7 g/dL. No patients required transfusions following OMS906 treatment. Mean absolute reticulocyte counts were reduced by 70,000/μL–136,000/μL at all timepoints. OMS906 was well tolerated.
-
The most common adverse events were iron deficiency and transient neutropenia. No patients had clinical breakthrough hemolysis. Two patients had increases in LDH and bilirubin, suggesting initiation of hemolysis at the end of a dosing period, although Hgb was not reduced in either.
-
In terms of safety, it appeared to be well tolerated, with 2 patients reporting headache and 3 reporting itching, but there were no major TEAEs. Its efficacy also appeared promising, as all patients without myelodysplastic syndrome reached hemoglobin levels considered normal for their gender.
Summary
In this interim analysis of a proof-of-concept study of MASP-3 inhibition, once-monthly SC OMS906 in treatment naïve PNH patients demonstrated clinically meaningful increases in hemoglobin, reduction in LDH, transfusion independence, and reticulocyte reduction. OMS906 treatment was well tolerated with no safety signals of concern. The dose-timing–related biochemical changes observed in the 2 patients noted above will be used to guide future dose escalation. OMS906 dose escalation guided by the [pharmacokinetics and pharmacodynamics] of patients experiencing subclinical hemolysis is underway to inform achievement of quarterly dosing.
Reference: European Hematology Association 2023 Hybrid Congress, 8th-11th June 2023, Frankfurt, Germany.