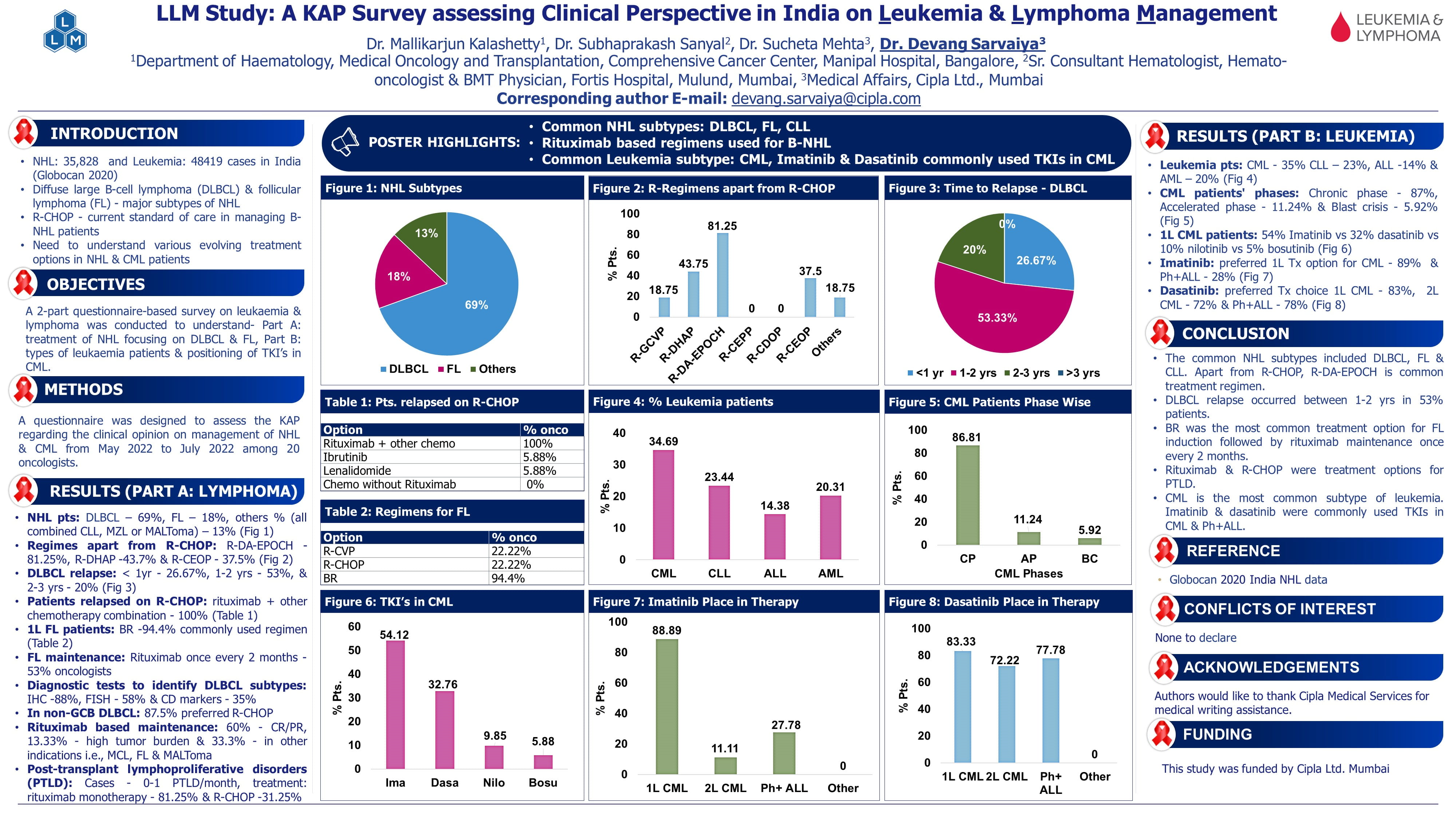
Individualized, PK-Guided Dosing of Hydroxyurea for Young Children with Sickle Cell Anemia: Final Results from the Hydroxyurea Optimization through Precision Study (HOPS)
Speaker: Abena Appiah-Kubi, Zucker SOM, Hofstra/Northwell
Key Highlights:
Introduction:
Hydroxyurea, recommended for children with sickle cell anemia by the National Heart, Lung and Blood Institute (NHLBI) guidelines, has shown substantial benefits in increasing fetal hemoglobin (HbF). However, concerns about elevated baseline HbF levels in infants and time-consuming dose escalation processes have limited its widespread adoption. Pharmacokinetics (PK)-guided dosing presents a tailored approach, potentially reducing the time required to achieve optimal outcomes.
Previous studies have shown that the effective hydroxyurea doses range from 15-35 mg/kg/day. The pharmacokinetics and the HbF response vary from individual to individual.
HOPS Study Design:
Dr. Abena Appiah-Kubi presented findings from the Hydroxyurea Optimization through Precision Study (HOPS), phase 3, randomized, prospective clinical study conducted across 14 pediatric sickle cell centers in the United States.
The objective was to compare PK-guided hydroxyurea dosing with the standard fixed dosing regimen of 20 mg/kg/day.
- Participants:
- Age range: 6 months to 21 years
- Total enrolled: 127
- 13 withdrew before starting hydroxyurea
- 10 withdrew before month 6 (mostly due to loss to follow-up).
- 104 completed the study and reached the primary endpoint.
- Primary Endpoint: HbF levels at 6 months
- Dosing:
- Standard arm: Fixed 20 mg/kg/day dose
- PK-guided arm: Starting doses adjusted to achieve target AUC (e.g., adjusted from 20 mg/kg to 28 mg/kg based on PK data)
- Dose escalation and adjustment criteria were consistent for both arms, considering growth-related weight changes in participants
- Dose Holding Criteria:
- Absolute Neutrophil Count (ANC) < 750/µL.
- Absolute Reticulocyte Count < 100 × 10⁹/L if hemoglobin < 8 g/dL.
- Platelets < 80,000.
Baseline Characteristics:
- Age (median, IQR): 10 months (7.1-17.5) in PK-Guided vs. 8 months (7.3-22.8) in Weight-Based.
- WBC (10³/L): 10.4 ± 3.2 in PK-Guided vs. 10.8 ± 4.6 in Weight-Based.
- ANC (10³/L): 3.1 ± 1.8 in PK-Guided vs. 3.2 ± 2.0 in Weight-Based.
- Hemoglobin (g/dL): 9.0 ± 1.1 in PK-Guided vs. 8.9 ± 1.2 in Weight-Based.
- ARC (10³/L): 274 ± 120 in PK-Guided vs. 256 ± 127 in Weight-Based.
- HbF (%): 27.0 ± 11.4 in PK-Guided vs. 27.7 ± 11.2 in Weight-Based.
Results:
- Rapid Optimization and Hematologic Benefits:
- The PK-guided arm started with significantly higher doses (average 27.5 mg/kg/day) than the weight-based arm (20 mg/kg/day).
- PK dosing achieved higher HbF increases (mean delta of 10% vs. 5%) at six months.
- Both groups exceeded 30% HbF by six months, with over 70% F-cells in both cohorts, indicating a pan cellular HbF distribution.
- Safety and Tolerability:
- Both arms demonstrated comparable safety profiles, with no significant differences in hematologic toxicity. There were 43 dose holds equally distributed between the groups.
- Only five participants required dose reductions during the study.
- Practical Considerations: PK-guided dosing eliminated the need for prolonged dose escalation, which took up to a year in the weight-based group to reach similar efficacy.
Conclusion:
Dr. Abena concluded, PK-guided dosing of hydroxyurea resulted in a higher HbF increase by month 6 and provided the optimal dose from the start, whereas weight-based dosing took up to a year to achieve similar levels. The strategy, with adjustments for weight gain, was safe and led to significant hematologic improvements in very young children. The HOPS results suggest that early initiation of hydroxyurea at a dose of 25 mg/kg/day is both feasible and effective.
A Phase 1b/2 Study of the Efficacy and Safety of Combination Hydroxyurea and Dose-Escalated Erythropoietin for Treatment of Anaemia in Sickle Cell Disease (ACHiEvE-SCD)
Speaker: Julia Xu, University of Pittsburgh
Key Highlights:
Study Rationale:
Chronic anaemia in Sickle Cell Disease (SCD) exacerbates organ ischemia, inflammation, and cardiovascular complications.
Hydroxyurea, a mainstay therapy, provides modest haemoglobin increases in many patients. While EPO is effective for anaemia in chronic kidney disease, it is underutilized in SCD due to mixed efficacy, safety concerns (e.g., thrombosis, vaso-occlusive crises), and a lack of standardized dosing guidelines.
Study Details:
Dr. Xu explained the ACHiEvE-SCD study (NCT05451940).
- Study Objective: Evaluate the efficacy and safety of epoetin alpha in sickle cell patients with anaemia.
- Sites: University of Pittsburgh and Lagos University Teaching Hospital, Nigeria (high global burden of SCD).
Study Design:
- Phase: 1b/2 Open-label study
- Population:
- Adults with HbSS or S beta-0 thalassemia
- On stable hydroxyurea with Hb ≤ 9 g/dL
- No recent transfusions, Voxelotor, or ESA use
- Excluded: Dialysis patients or recent vaso-occlusive crises
- Treatment Regimen:
- Duration: 12 weeks of dose-escalated epoetin alpha, followed by a 12-week observation period.
- Starting Dose: 10,000 units, 3 times/week, escalated up to 40,000 units as needed to target Hb ≥ 10 g/dL.
- Dose Adjustments: Hb > 11 g/dL required dose modification.
- Primary Endpoint: Hb response — defined as an increase of ≥1 g/dL from baseline at 12 weeks.
- Key Metrics:
- Screening and Enrolment:
- 23 subjects screened
- 6 excluded (ineligibility/lost to follow-up)
- Screening and Enrolment:
- 17 enrolled and dosed
- Completion:
- 16 patients completed the study
- 1 patient withdrawn due to unrelated serious adverse event
- 15 completed treatments; 1 remains in active treatment.
- Monitoring:
- Lab markers (safety and titration): Every 2 weeks
- Clinical and patient-reported outcomes: Baseline, 12 weeks, 24 weeks
Findings: Baseline: Mean Hb 7.4 g/dL; mean hydroxyurea dose 22 mg/kg/day.
Efficacy:
- 93% (13/14) achieved ≥1 g/dL Hb increase at 12 weeks.
- Mean Hb increase of 3.1 g/dL at 12 weeks; 1 g/dL increase achieved by week 2.
- Significant reductions in hemolytic markers (LDH, bilirubin, reticulocytes) and ferritin.
- HbF increased by 2.6%.
- Subgroup Analysis
- Response was robust in both CKD and non-CKD patients, with slightly higher increases observed in those with CKD.
- Average effective EPO dose: 20,000 units 3 times per week.
Safety:
- Adverse events were mild to moderate; no events directly related to EPO.
- Two serious adverse events: one patient developed myelodysplastic syndrome (MDS, unrelated to treatment), and one had a vaso-occlusive crisis.
- Compliance was high, with 97% adherence.
Conclusion:
Dr. Xu concluded,
- In summary, anaemia contributes to chronic organ damage and reduced quality of life in SCD. Our study utilized standardized epoetin alpha dosing to treat anaemia in patients stabilized on hydroxyurea.
- Key Findings:
- Robust Hb Response: Achieved within 12 weeks of treatment.
- Safety: Demonstrated an acceptable safety profile.
- Future analyses will explore correlations with neurocognitive function, cardiopulmonary function, and pain experience and additional studies will examine blood rheology and tissue oxygenation via near-infrared spectroscopy.
Etavopivat Reduces Incidence of Vaso-Occlusive Crises in Patients with Sickle Cell Disease: HIBISCUS Trial Phase 2 Results through 52 Weeks
Speaker: Sophia Delicou, Hippokrateio General Hospital
Key Highlights:
Introduction:
Etavopivat is a selective, once-daily oral activator of pyruvate kinase in red blood cells. Through allosteric activation, Etavopivat enhances ATP production and reduces 2,3-DPG concentrations, leading to the stabilization of red blood cell membranes and a reduction in Hb S polymerization—all without impairing oxygen delivery.
In a Phase 1 study, Etavopivat demonstrated promising results in patients with SCD, showing a rapid and sustained increase in Hb levels over 12 weeks of treatment while also reducing markers of hemolysis.
Study Design:
- Type: Multi-centre, double-blind, placebo-controlled trial.
- Population: Patients aged 12–65 years with all SCD genotypes, Hb levels of 5.5–10.5 g/dL, and 2–10 VOCs requiring medical care in the past year.
- Intervention: Randomized to receive 200 mg, 400 mg of etavopivat, or placebo daily for 52 weeks, followed by an optional 112-week open-label extension.
- Primary Endpoints:
- Annualized VOC rate over 52 weeks.
- Hb response (≥1 g/dL increase from baseline) at week 24.
- Secondary Endpoints: Hemolysis markers, time to first VOC, fatigue (PROMIS scale), and safety.
Key Results:
Efficacy in Reducing VOCs:
- Intent-to-Treat Analysis:
- 200 mg: 45% reduction in annualized VOC rate vs. placebo.
- 400 mg: 46% reduction vs. placebo.
- Per-Protocol Analysis:
- 200 mg: 63% reduction in VOCs.
- 400 mg: 61% reduction in VOCs.
- Median time to first VOC was significantly extended (8 months for etavopivat vs. 4 months for placebo).
Hb Response:
- Both doses achieved early haemoglobin increases by week 2, sustained through 52 weeks.
- Mean baseline haemoglobin: ~8.5 g/dL.
- Week 24: Significant increases observed in both intent-to-treat and per-protocol groups.
Haemolysis Markers:
- LDH and bilirubin levels decreased significantly by week 24 and remained stable through week 52.
- LDH proved to be a valuable biomarker for haemolysis monitoring.
Quality of Life: Fatigue scores improved (PROMIS scale), indicating better patient-reported outcomes.
Safety Profile
- Etavopivat was well tolerated, with most adverse events (AEs) being mild to moderate.
- Common AEs included nausea, headache, urinary tract infections, and insomnia, predominantly unrelated to the drug.
- Serious AEs (e.g., cerebrovascular accident, elevated hepatic enzymes) occurred in a few patients and led to discontinuation in 2 cases but were not considered drug related.
- No deaths were reported during the trial.
Conclusion:
- Based on pharmacokinetics, pharmacodynamics, and dose-response analysis, the 400 mg/day dosage of etavopivat was selected for confirmatory Phase 3 trials.
- Etavopivat was well-tolerated with no unexpected safety concerns identified.
- Compared to placebo, daily etavopivat use in the intent-to-treat population showed positive trends, including:
- Lower annualized VOC rates through week 52.
- Delayed time to the first VOC.
- Early increases in Hb levels by week 2 and higher Hb response rates by week 24.
- Reduced fatigue scores by week 52.
- Decreased markers of hemolysis.
- These findings support improved Hb oxygen affinity and enhanced red blood cell health in SCD with oral etavopivat.
ASH Annual Meeting and Exposition, 7-10 December 2024, San Diego, California